A Hipoplasia Pontocerebelar tipo 7 (HPC7) é uma forma muito rara e recém-descoberta dessa condição, com causa desconhecida e um prognóstico desfavorável (futuro ruim). Até o momento, foi relatada em apenas quatro pacientes. Ela é identificada logo após o nascimento (período neonatal) por fraqueza muscular generalizada (hipotonia), ausência de gônadas (órgãos sexuais como testículos ou ovários) que possam ser sentidas ao toque, e micropênis (pênis menor que o normal). A partir da infância (poucos meses de idade), as características incluem microcefalia progressiva (a cabeça é pequena e seu crescimento não acompanha o esperado), episódios de apneia (paradas breves da respiração), dificuldade para se alimentar, convulsões e até mesmo uma diminuição do tamanho do pênis. Exames de imagem, como a ressonância magnética, mostram que partes do cérebro chamadas ponte e cerebelo não se desenvolveram completamente (uma condição chamada hipoplasia pontocerebelar). Em pessoas com cromossomos XY (ou seja, geneticamente homens), a HPC7 se manifesta como a hipoplasia pontocerebelar junto com um distúrbio no desenvolvimento sexual (problemas na formação dos órgãos sexuais). Já em pessoas com cromossomos XX (geneticamente mulheres), ela pode se manifestar apenas como a hipoplasia pontocerebelar, sem os problemas evidentes de desenvolvimento sexual.
Introdução
O que você precisa saber de cara
A Hipoplasia Pontocerebelar tipo 7 (HPC7) é uma forma muito rara e recém-descoberta dessa condição, com causa desconhecida e um prognóstico desfavorável (futuro ruim). Até o momento, foi relatada em apenas quatro pacientes. Ela é identificada logo após o nascimento (período neonatal) por fraqueza muscular generalizada (hipotonia), ausência de gônadas (órgãos sexuais como testículos ou ovários) que possam ser sentidas ao toque, e micropênis (pênis menor que o normal). A partir da infância (poucos meses de idade), as características incluem microcefalia progressiva (a cabeça é pequena e seu crescimento não acompanha o esperado), episódios de apneia (paradas breves da respiração), dificuldade para se alimentar, convulsões e até mesmo uma diminuição do tamanho do pênis. Exames de imagem, como a ressonância magnética, mostram que partes do cérebro chamadas ponte e cerebelo não se desenvolveram completamente (uma condição chamada hipoplasia pontocerebelar). Em pessoas com cromossomos XY (ou seja, geneticamente homens), a HPC7 se manifesta como a hipoplasia pontocerebelar junto com um distúrbio no desenvolvimento sexual (problemas na formação dos órgãos sexuais). Já em pessoas com cromossomos XX (geneticamente mulheres), ela pode se manifestar apenas como a hipoplasia pontocerebelar, sem os problemas evidentes de desenvolvimento sexual.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 25 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 66 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisInhibits cell growth rate and cell cycle. Induces CDKN1A expression as well as TGF-beta expression. Mediates the inhibitory growth effect of EGR1. Involved in the maturation of snRNAs and snRNA 3'-tail processing (PubMed:28092684)
Nucleus, nucleolusNucleus speckle
Pontocerebellar hypoplasia 7
A form of pontocerebellar hypoplasia, a group of related disorders characterized by underdevelopment of the pons and the cerebellum. Pontocerebellar hypoplasia also causes impaired growth of other parts of the brain, leading to an unusually small head size. PCH7 patients manifest delayed psychomotor development, hypotonia, breathing abnormalities, and gonadal abnormalities.
Multiple inositol polyphosphate phosphatase that hydrolyzes 1D-myo-inositol 1,3,4,5,6-pentakisphosphate (InsP5[2OH]) and 1D-myo-inositol hexakisphosphate (InsP6) to a range of less phosphorylated inositol phosphates. This regulates the availability of these various small molecule second messengers and metal chelators which control many aspects of cell physiology (PubMed:33257696, PubMed:36589890). Has a weak in vitro activity towards 1D-myo-inositol 1,4,5-trisphosphate which is unlikely to be ph
Endoplasmic reticulum lumenSecretedCell membrane
Thyroid cancer, non-medullary, 2
A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms.
Variantes genéticas (ClinVar)
213 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 37 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipoplasia pontocerebelar tipo 7
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
46,XY differences of sex development in pontocerebellar hypoplasia type 7 (PCH7): two case reports and systematic review.
We report two cases of 46,XY siblings with pontocerebellar hypoplasia type 7 (PCH7) and conduct a systematic literature review for genetically confirmed PCH7 cases, focusing on phenotypic characteristics, particularly gonadal parameters, and associated genotypic data. Two 46,XY siblings diagnosed with PCH7 were reviewed. Both exhibited hypoplastic male external genitalia, absent uterus, and cryptorchid testes, confirmed through histological assessments showing dysgenesis. A systematic literature search revealed an additional 26 cases of 46,XY PCH7, with neurological involvement noted in all cases except one. The external genitalia were described as abnormal (17/23); however, few of these were hypoplastic male type on pictorial review. Nonlocalized testes (5/5) and absent uterus (4/7) on ultrasonography, elevated FSH (7/7), and low testosterone (3/3) were observed. Besides a novel variant (p.Ile133Thr) in Indian siblings, a total of 25 variants in TOE1 were identified with no specific genotype-phenotype correlation. Testicular development was defective in all PCH7 patients but was variable, with the predominant phenotypic manifestation being testicular regression syndrome/partial gonadal dysgenesis with Müllerian duct regression (TRS/PGD-MDR).
Biochemical characterizations of Pontocerebellar Hypoplasia linked mutations of Target of Egr1 (TOE1) reveal impacts on thermal stability, ribonuclease activity, and oligomerization.
The Target of EGR1 (TOE1) gene encodes the TOE1 deadenylase, which is essential for the maturation of Pol-II transcribed snRNAs in humans. Over a dozen missense mutations in the TOE1 gene have been linked to Pontocerebellar Hypoplasia Type 7 (PCH7), a rare but serious neurodevelopmental and neurodegenerative disease that leads to early mortality. The biochemical mechanisms for why these PCH7-linked mutations alter TOE1's biochemical characteristics remains vague. Here, we utilized AlphaFold predicted structures of TOE1 and biochemical characterizations to investigate the impact of TOE1 variants on TOE1's biochemical properties. We performed characterization of the thermal stability and activity of eleven PCH-linked TOE1 variants and found that eight variants have significant reduced protein thermal stability and only two variants impair TOE1's ribonuclease activity, particularly its exonuclease activity. Additionally, we found that the F148Y mutation impacts TOE1's oligomeric state in vitro and in vivo. Together, these results demonstrated that PCH-linked mutations of TOE1 impact many different aspects of TOE1 biochemistry, providing novel insights which may provide potential therapeutic strategies to treat PCH7 patients. In addition, these mutations provide a library of TOE1 variants that will be useful for future studies of TOE1 function and regulation.
Novel heterozygous missense variants in the TOE1 gene linked to pontocerebellar hypoplasia type 7.
Role of TOE1 variants at the nuclear localization motif in pontocerebellar hypoplasia 7.
Biallelic TOE1 variants can cause pontocerebellar hypoplasia type 7 (PCH7), a condition characterized by pontocerebellar hypoplasia with genital abnormality. TOE1 is a 3'-exonuclese for 3'-end maturation in small nuclear RNA. TOE1 pathogenic variants have been reported at the DEDD catalytic domain and zinc finger motif. Here, we describe a PCH7 patient with novel compound heterozygous TOE1 variants and a detailed clinical course. The patient was a 3-year-old female and showed developmental delay without cerebellar ataxic behavior. Head MRI revealed delayed myelination without pontocerebellar hypoplasia at 9 months of age. Progressive pontocerebellar atrophy was prominent at follow-up MRI. Cerebral abnormalities are characteristic features of PCH7 before pontocerebellar atrophy is observed. One variant, p.Arg331*, was located at the nuclear localization motif (NLM) and partially escaped from nonsense-mediated decay. This variant affected nuclear localization in mutant expressing cells, thus, the TOE1 variant at NLM leads to TOE1 dysfunction associated with nuclear mis-localization.
Case report: A severe clinical phenotype of pontocerebellar hypoplasia type 7 with compound heterozygous variants of TOE1.
Pontocerebellar Hypoplasia (PCH) is a rare autosomal recessive hereditary neurological degenerative disease. To elaborate upon the clinical phenotypes of PCH and explore the correlation between TOE1 gene mutations and clinical phenotype, we analyze the clinical and genetic features of a Chinese infant afflicted with pontocerebellar dysplasia accompanied by gender reversal with bioinformatics methods. The main clinical features of this infant with TOE1 gene mutation included progressive lateral ventricle widening, hydrocephalus, severe postnatal growth retardation, and hypotonia, and simultaneously being accompanied by 46, XY female sex reversal. Whole exome sequencing revealed a compound heterozygous mutation in the TOE1 gene (c.299T > G, c.1414T > G), with the protein homology modeling-generated structure predicting a pathogenic variation, which is closely related to the clinical manifestations in the patient. The new mutation sites, c.299T > G and c.1414T > G, in the TOE1 gene are pathogenic variants of pontocerebellar hypoplasia type 7.
Publicações recentes
46,XY differences of sex development in pontocerebellar hypoplasia type 7 (PCH7): two case reports and systematic review.
Biochemical characterizations of Pontocerebellar Hypoplasia linked mutations of Target of Egr1 (TOE1) reveal impacts on thermal stability, ribonuclease activity, and oligomerization.
Novel heterozygous missense variants in the TOE1 gene linked to pontocerebellar hypoplasia type 7.
Role of TOE1 variants at the nuclear localization motif in pontocerebellar hypoplasia 7.
Case report: A severe clinical phenotype of pontocerebellar hypoplasia type 7 with compound heterozygous variants of TOE1.
📚 EuropePMC251 artigos no totalmostrando 12
46,XY differences of sex development in pontocerebellar hypoplasia type 7 (PCH7): two case reports and systematic review.
Journal of pediatric endocrinology & metabolism : JPEMBiochemical characterizations of Pontocerebellar Hypoplasia linked mutations of Target of Egr1 (TOE1) reveal impacts on thermal stability, ribonuclease activity, and oligomerization.
bioRxiv : the preprint server for biologyNovel heterozygous missense variants in the TOE1 gene linked to pontocerebellar hypoplasia type 7.
Genes & diseasesRole of TOE1 variants at the nuclear localization motif in pontocerebellar hypoplasia 7.
Journal of human geneticsCase report: A severe clinical phenotype of pontocerebellar hypoplasia type 7 with compound heterozygous variants of TOE1.
HeliyonGenetic and prenatal diagnosis of a Chinese pedigree with pathogenic TOE1 variants causing pontocerebellar hypoplasia type 7.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansClinical and genetic characterization of a Chinese family with pontocerebellar hypoplasia type 7.
American journal of medical genetics. Part AThe PARN, TOE1, and USB1 RNA deadenylases and their roles in non-coding RNA regulation.
The Journal of biological chemistryNovel compound heterozygous missense variants in TOE1 gene associated with pontocerebellar hypoplasia type 7.
GeneKnockdown of Toe1 causes developmental arrest during the morula-to-blastocyst transition in mice.
TheriogenologyNovel compound heterozygous variant of TOE1 results in a mild type of pontocerebellar hypoplasia type 7: an expansion of the clinical phenotype.
NeurogeneticsBiallelic mutations in the 3' exonuclease TOE1 cause pontocerebellar hypoplasia and uncover a role in snRNA processing.
Nature geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipoplasia pontocerebelar tipo 7.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipoplasia pontocerebelar tipo 7
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- 46,XY differences of sex development in pontocerebellar hypoplasia type 7 (PCH7): two case reports and systematic review.
- Biochemical characterizations of Pontocerebellar Hypoplasia linked mutations of Target of Egr1 (TOE1) reveal impacts on thermal stability, ribonuclease activity, and oligomerization.
- Novel heterozygous missense variants in the TOE1 gene linked to pontocerebellar hypoplasia type 7.
- Role of TOE1 variants at the nuclear localization motif in pontocerebellar hypoplasia 7.
- Case report: A severe clinical phenotype of pontocerebellar hypoplasia type 7 with compound heterozygous variants of TOE1.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:284339(Orphanet)
- OMIM OMIM:614969(OMIM)
- MONDO:0013993(MONDO)
- GARD:17315(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18966160(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
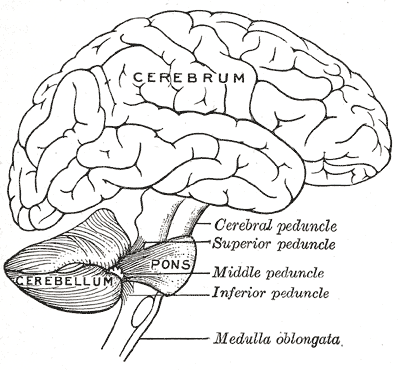
Hipoplasia pontocerebelar tipo 7
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata