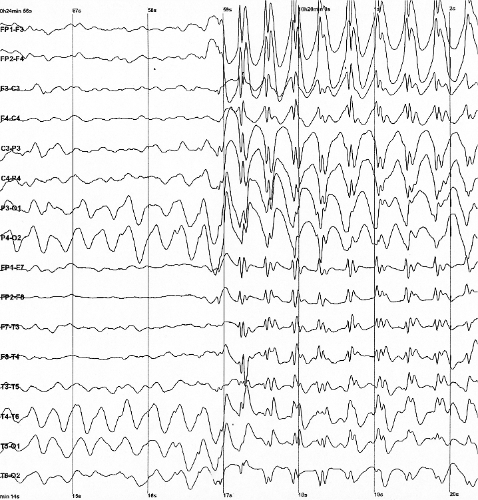
Síndrome epiléptica infantil rara, caracterizada pelo início precoce de convulsões de tipo e gravidade variáveis, potencialmente associadas a um espectro de sinais e sintomas clínicos, incluindo atraso ou falta de desenvolvimento psicomotor, deficiência intelectual, desenvolvimento deficiente ou ausente da fala, anormalidades comportamentais, hipotonia, distúrbios motores, espasticidade, microcefalia e características faciais dismórficas, entre outros. Os achados de imagem cerebral também são variáveis e podem incluir atrofia cerebral ou anomalias da substância branca.
Introdução
O que você precisa saber de cara
Síndrome epiléptica infantil rara, caracterizada pelo início precoce de convulsões de tipo e gravidade variáveis, potencialmente associadas a um espectro de sinais e sintomas clínicos, incluindo atraso ou falta de desenvolvimento psicomotor, deficiência intelectual, desenvolvimento deficiente ou ausente da fala, anormalidades comportamentais, hipotonia, distúrbios motores, espasticidade, microcefalia e características faciais dismórficas, entre outros. Os achados de imagem cerebral também são variáveis e podem incluir atrofia cerebral ou anomalias da substância branca.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 57 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 159 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
55 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, Not applicable, X-linked recessive.
Curadoria gene-doença
fontes oficiaisInvolved in the regulation of endosome-to-lysosome trafficking, including endocytic trafficking of EGF-EGFR complexes and GABA-A receptors (PubMed:18675823). Involved in mitochondrial motility. When O-glycosylated, abolishes mitochondrial motility. Crucial for recruiting OGT to the mitochondrial surface of neuronal processes (PubMed:24995978). TRAK1 and RHOT form an essential protein complex that links KIF5 to mitochondria for light chain-independent, anterograde transport of mitochondria (By si
CytoplasmNucleusMitochondrionEarly endosomeEndosomeMitochondrion membraneCytoplasm, cell cortex
Developmental and epileptic encephalopathy 68
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE68 is an autosomal recessive form characterized by onset of twitching and/or myoclonic jerks in infancy. The disorder progresses to refractory generalized tonic-clonic seizures, often resulting in status epilepticus, loss of developmental milestones, and early death. Other features include delayed development, axial hypotonia, spasticity of the limbs, and clonus.
Putative oxidoreductase. Acts as a tumor suppressor and plays a role in apoptosis. Required for normal bone development (By similarity). May function synergistically with p53/TP53 to control genotoxic stress-induced cell death. Plays a role in TGFB1 signaling and TGFB1-mediated cell death. May also play a role in tumor necrosis factor (TNF)-mediated cell death. Inhibits Wnt signaling, probably by sequestering DVL2 in the cytoplasm
CytoplasmNucleusMitochondrionGolgi apparatusLysosome
Beta subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:10449790, PubMed:16412217, PubMed:26950270). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain one or two GABA active binding sites located at the alpha and beta subunit interfaces, depending on subunit composition (By similarity). When activated b
Postsynaptic cell membraneCell membrane
Developmental and epileptic encephalopathy 45
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Substrate-recognition component of the SCF (SKP1-CUL1-F-box protein)-type E3 ubiquitin ligase complex, promoting ubiquitination and proteasomal degradation of specific target proteins including TOP2A, RAB27A or itself (PubMed:27754753, PubMed:31678254). Regulates topoisomerase IIalpha/TOP2A decatenation activity and plays an important role in maintaining genomic stability (PubMed:27754753). Plays a role in lipid metabolism and inflammation through the ubiquitinated degradation of RAB27A (By simi
Chromosome, centromere, kinetochoreNucleusChromosome
Developmental and epileptic encephalopathy 100
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE100 is an autosomal dominant, severe form characterized by global developmental delay and onset of variable types of seizures in the first months or years of life.
Transcription repression factor which plays an important role in the establishment of the regional subdivision of the developing brain and in the development of the telencephalon
Nucleus
Rett syndrome congenital variant
A severe neurodevelopmental disorder with features of classic Rett syndrome but earlier onset in the first months of life. Clinical features include progressive microcephaly, hypotonia, irresponsiveness and irritability in the neonatal period, intellectual disability, psychomotor regression and stereotypical movements.
Voltage-gated potassium channel that mediates transmembrane potassium transport in excitable membranes, primarily in the brain. Contributes to the regulation of the fast action potential repolarization and in sustained high-frequency firing in neurons of the central nervous system. Homotetramer channels mediate delayed-rectifier voltage-dependent potassium currents that activate rapidly at high-threshold voltages and inactivate slowly. Forms tetrameric channels through which potassium ions pass
Cell membraneMembranePerikaryonCell projection, axonCell projection, dendritePostsynaptic cell membranePresynaptic cell membraneSynapse, synaptosomeSynapseApical cell membraneBasolateral cell membrane
With DHDDS, forms the dehydrodolichyl diphosphate synthase (DDS) complex, an essential component of the dolichol monophosphate (Dol-P) biosynthetic machinery (PubMed:21572394, PubMed:25066056, PubMed:28842490, PubMed:32817466, PubMed:33077723). Both subunits contribute to enzymatic activity, i.e. condensation of multiple copies of isopentenyl pyrophosphate (IPP) to farnesyl pyrophosphate (FPP) to produce dehydrodolichyl diphosphate (Dedol-PP), a precursor of dolichol phosphate which is utilized
Endoplasmic reticulum membrane
Congenital disorder of glycosylation 1AA
A form of congenital disorder of glycosylation, a multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. CDG1AA inheritance is autosomal recessive.
Pore-forming subunit of a voltage-gated sodium channel complex assuming opened or closed conformations in response to the voltage difference across membranes and through which sodium ions selectively pass along their electrochemical gradient (PubMed:24874546, PubMed:25239001, PubMed:25725044, PubMed:26900580, PubMed:29726066, PubMed:33245860, PubMed:36696443, PubMed:36823201). Contributes to neuronal excitability by regulating action potential threshold and propagation (PubMed:24874546, PubMed:2
Cell membraneCell projection, axonCytoplasmic vesicleCell projection, podosome
Cognitive impairment with or without cerebellar ataxia
A disorder characterized by markedly delayed cognitive and motor development, attention deficit disorder, and cerebellar ataxia. Features include bilateral esophoria, strabismatic amblyopia, unsustained gaze evoked nystagmus on horizontal gaze, ataxic gait, dysmetria in the upper limbs and dysarthria, with normal strength, tone, and reflexes.
Symporter that cotransports specific neutral amino acids and sodium ions, coupled to an H(+) antiporter activity (PubMed:10823827). Mainly participates in the glutamate-GABA-glutamine cycle in brain where it transports L-glutamine from astrocytes in the intercellular space for the replenishment of both neurotransmitters glutamate and gamma-aminobutyric acid (GABA) in neurons and also functions as the major influx transporter in ganglion cells mediating the uptake of glutamine (By similarity). Th
Cell membraneBasolateral cell membrane
Developmental and epileptic encephalopathy 102
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE102 is an autosomal recessive form characterized by onset of variable types of seizures in infancy.
Involved in T-cell adhesion and p53/TP53-dependent induction of apoptosis. Does not bind RNA. As component of the WAVE1 complex, required for BDNF-NTRK2 endocytic trafficking and signaling from early endosomes (By similarity)
CytoplasmNucleusCytoplasm, perinuclear regionSynapse, synaptosome
Developmental and epileptic encephalopathy 65
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE65 is an autosomal dominant form characterized by onset of intractable seizures usually in the first 6 months of life and severe to profound psychomotor developmental delay.
This is the catalytic component of the active enzyme, which catalyzes the hydrolysis of ATP coupled with the exchange of sodium and potassium ions across the plasma membrane. This action creates the electrochemical gradient of sodium and potassium, providing the energy for active transport of various nutrients
MembraneCell membrane
Migraine, familial hemiplegic, 2
A subtype of migraine with aura associated with hemiparesis in some families. Migraine is a disabling symptom complex of periodic headaches, usually temporal and unilateral. Headaches are often accompanied by irritability, nausea, vomiting and photophobia, preceded by constriction of the cranial arteries. Migraine with aura is characterized by recurrent attacks of reversible neurological symptoms (aura) that precede or accompany the headache. Aura may include a combination of sensory disturbances, such as blurred vision, hallucinations, vertigo, numbness and difficulty in concentrating and speaking.
High-affinity sodium/citrate cotransporter that mediates the entry of citrate into cells, which is a critical participant of biochemical pathways (PubMed:12445824, PubMed:12826022, PubMed:26324167, PubMed:26384929, PubMed:30054523, PubMed:33597751, PubMed:39622972). May function in various metabolic processes in which citrate has a critical role such as energy production (Krebs cycle), fatty acid synthesis, cholesterol synthesis, glycolysis, and gluconeogenesis (PubMed:12826022). Transports citr
Cell membrane
Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta
An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients.
Involved in transcriptional activation and repression of select genes by chromatin remodeling (alteration of DNA-nucleosome topology). Component of SWI/SNF chromatin remodeling complexes that carry out key enzymatic activities, changing chromatin structure by altering DNA-histone contacts within a nucleosome in an ATP-dependent manner. Belongs to the neuron-specific chromatin remodeling complex (nBAF complex), as such plays a role in remodeling mononucleosomes in an ATP-dependent fashion, and is
Nucleus
Developmental and epileptic encephalopathy 76
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE76 is an autosomal recessive form that may result in death in childhood.
With NUS1, forms the dehydrodolichyl diphosphate synthase (DDS) complex, an essential component of the dolichol monophosphate (Dol-P) biosynthetic machinery (PubMed:25066056, PubMed:28842490, PubMed:32817466, PubMed:33077723). Both subunits contribute to enzymatic activity, i.e. condensation of multiple copies of isopentenyl pyrophosphate (IPP) to farnesyl pyrophosphate (FPP) to produce dehydrodolichyl diphosphate (Dedol-PP), a precursor of dolichol phosphate which is utilized as a sugar carrier
Endoplasmic reticulum membrane
Retinitis pigmentosa 59
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Catalytic subunit of the V1 complex of vacuolar(H+)-ATPase (V-ATPase), a multisubunit enzyme composed of a peripheral complex (V1) that hydrolyzes ATP and a membrane integral complex (V0) that translocates protons (PubMed:8463241). V-ATPase is responsible for acidifying and maintaining the pH of intracellular compartments and in some cell types, is targeted to the plasma membrane, where it is responsible for acidifying the extracellular environment (PubMed:32001091). In aerobic conditions, invol
CytoplasmCytoplasm, cytosolCytoplasmic vesicle, secretory vesicleCytoplasmic vesicle, clathrin-coated vesicle membraneLysosome
Cutis laxa, autosomal recessive, 2D
A form of cutis laxa, a disorder characterized by an excessive congenital skin wrinkling, a large fontanelle with delayed closure, a typical facial appearance with downslanting palpebral fissures, and a general connective tissue weakness. Most ARCL2D patients exhibit severe hypotonia as well as cardiovascular and neurologic involvement.
Subunit of non-clathrin- and clathrin-associated adaptor protein complex 3 (AP-3) that plays a role in protein sorting in the late-Golgi/trans-Golgi network (TGN) and/or endosomes. The AP complexes mediate both the recruitment of clathrin to membranes and the recognition of sorting signals within the cytosolic tails of transmembrane cargo molecules. AP-3 appears to be involved in the sorting of a subset of transmembrane proteins targeted to lysosomes and lysosome-related organelles. In concert w
Cytoplasmic vesicle, clathrin-coated vesicle membraneGolgi apparatus
Developmental and epileptic encephalopathy 48
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE48 is an autosomal recessive form characterized by onset of seizures in the first year of life. Affected individuals manifest global developmental delay, intellectual disability, absent speech, and poor, if any, motor development.
Voltage-gated potassium channel that mediates transmembrane potassium transport in excitable membranes, primarily in the brain, but also in the pancreas and cardiovascular system. Contributes to the regulation of the action potential (AP) repolarization, duration and frequency of repetitive AP firing in neurons, muscle cells and endocrine cells and plays a role in homeostatic attenuation of electrical excitability throughout the brain (PubMed:23161216). Plays also a role in the regulation of exo
Cell membranePerikaryonCell projection, axonCell projection, dendriteMembranePostsynaptic cell membraneSynapseSynapse, synaptosomeLateral cell membraneCell membrane, sarcolemma
Developmental and epileptic encephalopathy 26
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE26 patients manifest multiple types of seizures, delayed psychomotor development, poor or absent speech, hypotonia, hypsarrhythmia.
May function as an adapter protein or regulator of Ras signaling pathways
CytoplasmMembrane
Intellectual developmental disorder, X-linked, syndromic, Houge type
A disorder characterized by delayed development, intellectual disability, speech and language delay, and early-onset seizures. Carrier females may be mildly affected.
Phosphatase that acts on various phosphoinositides, including phosphatidylinositol 4-phosphate, phosphatidylinositol (4,5)-bisphosphate and phosphatidylinositol (3,4,5)-trisphosphate (PubMed:23804563, PubMed:27435091). Has a role in clathrin-mediated endocytosis (By similarity). Hydrolyzes PIP2 bound to actin regulatory proteins resulting in the rearrangement of actin filaments downstream of tyrosine kinase and ASH/GRB2 (By similarity)
Cytoplasm, perinuclear region
Parkinson disease 20, early-onset
An early-onset form of Parkinson disease, a complex neurodegenerative disorder characterized by bradykinesia, resting tremor, muscular rigidity and postural instability, as well as by a clinically significant response to treatment with levodopa. The pathology involves the loss of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies (intraneuronal accumulations of aggregated proteins), in surviving neurons in various areas of the brain. PARK20 is characterized by young adult-onset of parkinsonism. Additional features may include seizures, cognitive decline, abnormal eye movements, and dystonia.
Involved in nervous system development and function. Involved in the positive regulation of voltage-gated sodium channel activity. Promotes neuronal excitability by elevating the voltage dependence of neuronal sodium channel SCN8A fast inactivation
Nucleus
Developmental and epileptic encephalopathy 47
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
E1-like enzyme which specifically catalyzes the first step in ufmylation (PubMed:15071506, PubMed:18442052, PubMed:20368332, PubMed:25219498, PubMed:26929408, PubMed:27545674, PubMed:27545681, PubMed:27653677, PubMed:30412706, PubMed:30626644, PubMed:34588452). Activates UFM1 by first adenylating its C-terminal glycine residue with ATP, and thereafter linking this residue to the side chain of a cysteine residue in E1, yielding a UFM1-E1 thioester and free AMP (PubMed:20368332, PubMed:26929408, P
CytoplasmNucleusEndoplasmic reticulum membraneGolgi apparatus
Developmental and epileptic encephalopathy 44
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE44 transmission pattern is consistent with autosomal recessive inheritance.
Involved in tRNA methylation. Facilitates the recognition and targeting of tRNA(Arg)(CCU) and tRNA(Arg)(UCU) substrates for N(3)-methylcytidine modification by METTL2A and METTL2B
Developmental and epileptic encephalopathy 86
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE86 inheritance is autosomal recessive.
Pore-forming subunit of Nav1.3, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membraneBasal cell membrane
Epilepsy, familial focal, with variable foci 4
An autosomal dominant form of epilepsy characterized by focal seizures arising from different cortical regions, including the temporal, frontal, parietal, and occipital lobes. Seizure types commonly include temporal lobe epilepsy, frontal lobe epilepsy, and nocturnal frontal lobe epilepsy. Some patients may have intellectual disability or autism spectrum disorders. Seizure onset usually occurs in the first or second decades, although later onset has been reported, and there is phenotypic variability within families. A subset of patients have structural brain abnormalities. Penetrance of the disorder is incomplete. FFEVF4 is characterized by onset of focal seizures in the first years of life.
Multifunctional sorting protein that controls the endoplasmic reticulum (ER)-mitochondria communication, including the apposition of mitochondria with the ER and ER homeostasis. In addition, in response to apoptotic inducer, translocates BIB to mitochondria, which initiates a sequence of events including the formation of mitochondrial truncated BID, the release of cytochrome c, the activation of caspase-3 thereby causing cell death. May also be involved in ion channel trafficking, directing acid
Endoplasmic reticulumMitochondrion
Developmental and epileptic encephalopathy 66
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE66 is an autosomal dominant form characterized by onset of seizures in first days or weeks of life.
Involved in endocytosis
Cytoplasmic vesicle, clathrin-coated vesicle membraneCell membrane
Developmental and epileptic encephalopathy 21
A severe disease characterized by intractable seizures, profound global developmental delay, and persistent severe axial hypotonia as well as appendicular hypertonia.
Beta subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:19763268, PubMed:27789573, PubMed:29950725, PubMed:8264558). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit interface(s) (PubMed:29950725). When activated by GABA, GABAARs selecti
Postsynaptic cell membraneCell membraneCytoplasmic vesicle membrane
Epileptic encephalopathy, infantile or early childhood, 2
A form of epileptic encephalopathy, a heterogeneous group of severe childhood onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. IECEE2 is an autosomal dominant condition with variable age at seizure onset, ranging from early infancy to 6 years.
Component of N-methyl-D-aspartate (NMDA) receptors (NMDARs) that function as heterotetrameric, ligand-gated cation channels with high calcium permeability and voltage-dependent block by Mg(2+) (PubMed:26875626, PubMed:27616483, PubMed:28126851, PubMed:9489750). Participates in synaptic plasticity for learning and memory formation (By similarity). Channel activation requires binding of the neurotransmitter L-glutamate to the GluN2 subunit, glycine or D-serine binding to the GluN1 subunit, plus me
Cell membranePostsynaptic cell membrane
Developmental and epileptic encephalopathy 46
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Receptor tyrosine kinase involved in the development and the maturation of the central and the peripheral nervous systems through regulation of neuron survival, proliferation, migration, differentiation, and synapse formation and plasticity (By similarity). Receptor for BDNF/brain-derived neurotrophic factor and NTF4/neurotrophin-4. Alternatively can also bind NTF3/neurotrophin-3 which is less efficient in activating the receptor but regulates neuron survival through NTRK2 (PubMed:15494731, PubM
Cell membraneEndosome membraneEarly endosome membraneCell projection, axonCell projection, dendriteCytoplasm, perinuclear regionPostsynaptic density
Developmental and epileptic encephalopathy 58
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE58 is an autosomal dominant condition characterized by onset of refractory seizures in the first days or months of life.
Substrate-specific adapter for the anaphase promoting complex/cyclosome (APC/C) E3 ubiquitin-protein ligase complex. Associates with the APC/C in late mitosis, in replacement of CDC20, and activates the APC/C during anaphase and telophase. The APC/C remains active in degrading substrates to ensure that positive regulators of the cell cycle do not accumulate prematurely. At the G1/S transition FZR1 is phosphorylated, leading to its dissociation from the APC/C. Following DNA damage, it is required
NucleusCytoplasm
Developmental and epileptic encephalopathy 109
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE109 is an autosomal dominant form characterized by the onset of various types of seizures in the first months or years of life.
Hyperpolarization-activated ion channel that are permeable to sodium and potassium ions (PubMed:15351778, PubMed:28086084). Displays lower selectivity for K(+) over Na(+) ions (PubMed:28086084). Contributes to the native pacemaker currents in heart (If) and in the generation of the I(h) current which controls neuron excitability (PubMed:29936235, PubMed:30351409). Participates in cerebellar mechanisms of motor learning (By similarity). May mediate responses to sour stimuli (By similarity)
Cell membrane
Developmental and epileptic encephalopathy 24
A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals.
Alpha subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:14993607, PubMed:29961870, PubMed:30140029, PubMed:31056671). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit interface(s) (PubMed:30140029). When activated by GABA, GABAARs selec
Postsynaptic cell membraneCell membrane
Developmental and epileptic encephalopathy 79
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE79 is an autosomal dominant form characterized by onset of refractory seizures in the first months of life. Brain imaging may show hypomyelination, cerebral atrophy and thinning of the corpus callosum.
Component of a heterodimeric G-protein coupled receptor for GABA, formed by GABBR1 and GABBR2 (PubMed:15617512, PubMed:18165688, PubMed:22660477, PubMed:24305054, PubMed:9872316, PubMed:9872744). Within the heterodimeric GABA receptor, only GABBR1 seems to bind agonists, while GABBR2 mediates coupling to G proteins (PubMed:18165688). Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream e
Cell membranePostsynaptic cell membrane
Neurodevelopmental disorder with poor language and loss of hand skills
An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability.
Thiol-dependent isopeptidase that specifically cleaves UFM1, a ubiquitin-like modifier protein, from conjugated proteins, such as CD274/PD-L1, CYB5R3, DDRGK1, MRE11, RPL26/uL24, TRIP4 and RPL26/uL24 (PubMed:25219498, PubMed:27351204, PubMed:27926783, PubMed:30783677, PubMed:31595041, PubMed:32160526, PubMed:33473208, PubMed:35394863, PubMed:35926457, PubMed:36543799, PubMed:36893266, PubMed:37795761, PubMed:38383785). While it is also able to mediate the processing of UFM1 precursors, a prerequi
Endoplasmic reticulumCytoplasmNucleus
Beukes hip dysplasia
A severe progressive degenerative osteoarthritis of the hip joint with underlying dysplasia confined to that region. Affected individuals are of normal stature and have no associated health problems. Inheritance is autosomal dominant.
Sodium-dependent, high-affinity amino acid transporter that mediates the uptake of L-glutamate and also L-aspartate and D-aspartate (PubMed:14506254, PubMed:15265858, PubMed:26690923, PubMed:7521911). Functions as a symporter that transports one amino acid molecule together with two or three Na(+) ions and one proton, in parallel with the counter-transport of one K(+) ion (PubMed:14506254). Mediates Cl(-) flux that is not coupled to amino acid transport; this avoids the accumulation of negative
Cell membrane
Developmental and epileptic encephalopathy 41
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE41 inheritance is autosomal dominant.
Catalyzes the hydrolysis of GTP and utilizes this energy to mediate vesicle scission and participates in many forms of endocytosis, such as clathrin-mediated endocytosis or synaptic vesicle endocytosis as well as rapid endocytosis (RE) (PubMed:15703209, PubMed:20428113, PubMed:29668686, PubMed:8101525, PubMed:8910402, PubMed:9362482). Associates to the membrane, through lipid binding, and self-assembles into rings and stacks of interconnected rings through oligomerization to form a helical polym
Cell membraneMembrane, clathrin-coated pitCytoplasmic vesiclePresynapseCytoplasmic vesicle, secretory vesicle, chromaffin granule
Developmental and epileptic encephalopathy 31A
An autosomal dominant epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Calcium-dependent, calmodulin-stimulated protein phosphatase which plays an essential role in the transduction of intracellular Ca(2+)-mediated signals (PubMed:15671020, PubMed:18838687, PubMed:19154138, PubMed:23468591, PubMed:30254215). Many of the substrates contain a PxIxIT motif and/or a LxVP motif (PubMed:17498738, PubMed:17502104, PubMed:22343722, PubMed:23468591, PubMed:27974827). In response to increased Ca(2+) levels, dephosphorylates and activates phosphatase SSH1 which results in cof
CytoplasmCell membraneCell membrane, sarcolemmaCytoplasm, myofibril, sarcomere, Z lineCell projection, dendritic spine
Epileptic encephalopathy, infantile or early childhood, 1
A form of epileptic encephalopathy, a heterogeneous group of severe childhood onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. IECEE1 is an autosomal dominant condition with onset of seizures between the first weeks and first years of life.
This is the catalytic component of the active enzyme, which catalyzes the hydrolysis of ATP coupled with the exchange of sodium and potassium ions across the plasma membrane. This action creates the electrochemical gradient of sodium and potassium ions, providing the energy for active transport of various nutrients
Cell membrane
Dystonia 12
An autosomal dominant dystonia-parkinsonism disorder. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. DYT12 patients develop dystonia and parkinsonism between 15 and 45 years of age. The disease is characterized by an unusually rapid evolution of signs and symptoms. The sudden onset of symptoms over hours to a few weeks, often associated with physical or emotional stress, suggests a trigger initiating a nervous system insult resulting in permanent neurologic disability.
Alpha subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:10449790, PubMed:29961870, PubMed:31032849). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit interfaces (By similarity). When activated by GABA, GABAARs selectively allow the flow
Postsynaptic cell membraneCell membraneCytoplasmic vesicle membraneCell projection, dendrite
Developmental and epileptic encephalopathy 78
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE78 is an autosomal dominant form characterized by onset of refractory seizures in the first days or months of life. Clinical features include severe developmental delay, hypotonia, microcephaly, cortical visual impairment and profound intellectual disability. Some patients manifest a less severe phenotype characterized by pharmacoresponsive epilepsy, autism spectrum disorder and moderate intellectual disability.
RNA-binding protein implicated in the regulation of several post-transcriptional events. Involved in pre-mRNA alternative splicing, mRNA translation and stability. Mediates exon inclusion and/or exclusion in pre-mRNA that are subject to tissue-specific and developmentally regulated alternative splicing. Specifically activates exon 5 inclusion of TNNT2 in embryonic, but not adult, skeletal muscle. Activates TNNT2 exon 5 inclusion by antagonizing the repressive effect of PTB. Acts both as an activ
NucleusCytoplasm
Developmental and epileptic encephalopathy 97
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE97 is an autosomal dominant form.
Catalyzes the attachment of alanine to tRNA(Ala) in a two-step reaction: alanine is first activated by ATP to form Ala-AMP and then transferred to the acceptor end of tRNA(Ala) (PubMed:27622773, PubMed:27911835, PubMed:28493438, PubMed:33909043). Also edits incorrectly charged tRNA(Ala) via its editing domain (PubMed:27622773, PubMed:27911835, PubMed:28493438, PubMed:29273753). In presence of high levels of lactate, also acts as a protein lactyltransferase that mediates lactylation of lysine res
CytoplasmNucleus
Charcot-Marie-Tooth disease, axonal, type 2N
An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy.
As part of the KICSTOR complex functions in the amino acid-sensing branch of the TORC1 signaling pathway. Recruits, in an amino acid-independent manner, the GATOR1 complex to the lysosomal membranes and allows its interaction with GATOR2 and the RAG GTPases. Functions upstream of the RAG GTPases and is required to negatively regulate mTORC1 signaling in absence of amino acids. In absence of the KICSTOR complex mTORC1 is constitutively localized to the lysosome and activated. The KICSTOR complex
Lysosome membranePeroxisome
Developmental and epileptic encephalopathy 18
A severe autosomal recessive neurologic disorder characterized by lack of psychomotor development apparent from birth, dysmorphic facial features, early onset of refractory seizures, and thick corpus callosum and persistent cavum septum pellucidum on brain imaging.
Adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways (PubMed:15696159, PubMed:16511572, PubMed:36732624). Binds to a large number of partners, usually by recognition of a phosphoserine or phosphothreonine motif (PubMed:15696159, PubMed:16511572, PubMed:36732624). Binding generally results in the modulation of the activity of the binding partner (PubMed:16511572). Promotes inactivation of WDR24 component of the GATOR2 complex by bindi
Cytoplasm, cytosolMitochondrion matrix
Developmental and epileptic encephalopathy 56
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE56 is an autosomal dominant condition.
Voltage-sensitive calcium channels (VSCC) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. The isoform alpha-1A gives rise to P and/or Q-type calcium currents. P/Q-type calcium channels belong to the 'high-voltage activated' (HVA) group and are specifically blocked by the spider omega-agatoxin-IVA
Cell membrane
Spinocerebellar ataxia 6
Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA6 is an autosomal dominant cerebellar ataxia (ADCA), mainly caused by expansion of a CAG repeat in the coding region of CACNA1A. There seems to be a correlation between the repeat number and earlier onset of the disorder.
Voltage-sensitive calcium channels (VSCC) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. This alpha-1B subunit gives rise to N-type calcium currents. N-type calcium channels belong to the 'high-voltage activated' (HVA) group. They are involved in pain signaling (PubMed:25296916). Calcium channel
Membrane
Neurodevelopmental disorder with seizures and non-epileptic hyperkinetic movements
An autosomal recessive, complex and progressive neurologic disorder characterized by severe neurodevelopmental delay and developmental regression, epileptic encephalopathy, postnatal microcephaly, hypotonia, and non-epileptic hyperkinetic movement disorder, including myoclonus dystonia, choreoathetosis, or generalized dyskinesia. Disease onset in infancy or first years of life.
Translation elongation factor that catalyzes the GTP-dependent binding of aminoacyl-tRNA (aa-tRNA) to the A-site of ribosomes during the elongation phase of protein synthesis. Base pairing between the mRNA codon and the aa-tRNA anticodon promotes GTP hydrolysis, releasing the aa-tRNA from EEF1A1 and allowing its accommodation into the ribosome (By similarity). The growing protein chain is subsequently transferred from the P-site peptidyl tRNA to the A-site aa-tRNA, extending it by one amino acid
Endoplasmic reticulum membrane
Developmental and epileptic encephalopathy 33
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Voltage-gated potassium channel that mediates transmembrane potassium transport in excitable membranes, primarily in the brain and the central nervous system, but also in the cardiovascular system. Prevents aberrant action potential firing and regulates neuronal output. Forms tetrameric potassium-selective channels through which potassium ions pass in accordance with their electrochemical gradient. The channel alternates between opened and closed conformations in response to the voltage differen
Cell membraneMembraneCell projection, axonSynapseEndoplasmic reticulum membraneCell projection, lamellipodium membraneSynapse, synaptosomePresynaptic cell membraneCell projection, dendriteCell junction, paranodal septate junction
Developmental and epileptic encephalopathy 32
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE32 inheritance is autosomal dominant.
Pore-forming (alpha) subunit of a voltage-gated delayed rectifier potassium channel that mediates outward-rectifying potassium currents which, on depolarization, reaches a steady-state level and do not inactivate (PubMed:11943152, PubMed:12135768, PubMed:24133262, PubMed:36928654). The kinetic is characterized by a slow activation time course and a small voltage dependence of the activation time constants, therefore, starts to open at more negative voltages (PubMed:11943152, PubMed:12135768). Th
Membrane
Developmental and epileptic encephalopathy 112
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE112 is an autosomal dominant form characterized by onset in infancy, and a wide range of seizure types including focal and generalized seizures. Cognitive outcomes range from normal intellect to profound intellectual development impairment.
CytoplasmCytoplasm, perinuclear regionNucleus
Developmental and epileptic encephalopathy 87
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE87 inheritance is autosomal dominant.
Pore-forming subunit of Nav1.1, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membrane
Generalized epilepsy with febrile seizures plus 2
A rare autosomal dominant, familial condition with incomplete penetrance and large intrafamilial variability. Patients display febrile seizures persisting sometimes beyond the age of 6 years and/or a variety of afebrile seizure types. This disease combines febrile seizures, generalized seizures often precipitated by fever at age 6 years or more, and partial seizures, with a variable degree of severity.
Major constituent of the PSD essential for postsynaptic signaling. Inhibitory regulator of the Ras-cAMP pathway. Member of the NMDAR signaling complex in excitatory synapses, it may play a role in NMDAR-dependent control of AMPAR potentiation, AMPAR membrane trafficking and synaptic plasticity. Regulates AMPAR-mediated miniature excitatory postsynaptic currents. Exhibits dual GTPase-activating specificity for Ras and Rap. May be involved in certain forms of brain injury, leading to long-term lea
Intellectual developmental disorder, autosomal dominant 5
A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD5 patients show global developmental delay with delayed motor development, hypotonia, moderate-to-severe intellectual disability, and severe language impairment. Epilepsy and autism can be present in some patients.
Gamma subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:14993607, PubMed:16412217, PubMed:23909897, PubMed:2538761, PubMed:25489750, PubMed:27864268, PubMed:29950725, PubMed:30602789). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit in
Postsynaptic cell membraneCell membraneCell projection, dendriteCytoplasmic vesicle membrane
Developmental and epileptic encephalopathy 74
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE74 is an autosomal dominant form with onset in the first year of life.
As a component of the GATOR1 complex functions as an inhibitor of the amino acid-sensing branch of the mTORC1 pathway (PubMed:23723238, PubMed:25457612, PubMed:29590090, PubMed:29769719, PubMed:31548394, PubMed:35338845). In response to amino acid depletion, the GATOR1 complex has GTPase activating protein (GAP) activity and strongly increases GTP hydrolysis by RagA/RRAGA (or RagB/RRAGB) within heterodimeric Rag complexes, thereby turning them into their inactive GDP-bound form, releasing mTORC1
Lysosome membraneCytoplasm, cytosolCytoplasm, perinuclear region
Epilepsy, familial focal, with variable foci 1
An autosomal dominant form of epilepsy characterized by focal seizures arising from different cortical regions in different family members. Many patients have an aura and show automatisms during the seizures, whereas others may have nocturnal seizures. There is often secondary generalization. Some patients show abnormal interictal EEG, and some patients may have intellectual disability or autism spectrum disorders. Seizure onset usually occurs in the first or second decades, although later onset has been reported, and there is phenotypic variability within families. Penetrance of the disorder is incomplete.
Mitochondrial aminoacyl-tRNA synthetase that catalyzes the specific attachment of the proline amino acid (aa) to the homologous transfer RNA (tRNA), further participating in protein synthesis. The reaction occurs in a two steps: proline is first activated by ATP to form Pro-AMP and then transferred to the acceptor end of tRNA(Pro)
Mitochondrion matrix
Developmental and epileptic encephalopathy 75
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE75 is an autosomal recessive form characterized by onset of severe refractory seizures in the first months of life.
The alpha-2/delta subunit of voltage-dependent calcium channels regulates calcium current density and activation/inactivation kinetics of the calcium channel (PubMed:35293990). Plays an important role in excitation-contraction coupling (By similarity)
MembraneCell membrane
Developmental and epileptic encephalopathy 110
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE110 is an autosomal recessive form characterized by profound global developmental delay and hypotonia apparent in infancy followed by onset of seizures in the first months or years of life.
Clathrin is the major protein of the polyhedral coat of coated pits and vesicles. Two different adapter protein complexes link the clathrin lattice either to the plasma membrane or to the trans-Golgi network. Acts as a component of the TACC3/ch-TOG/clathrin complex proposed to contribute to stabilization of kinetochore fibers of the mitotic spindle by acting as inter-microtubule bridge (PubMed:15858577, PubMed:16968737, PubMed:21297582). The TACC3/ch-TOG/clathrin complex is required for the main
Cytoplasmic vesicle membraneMembrane, coated pitMelanosomeCytoplasm, cytoskeleton, spindle
Intellectual developmental disorder, autosomal dominant 56
A form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period.
Variantes genéticas (ClinVar)
405 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
143 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Encefalopatia epiléptica de início precoce inespecífica
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Variants loci and phenotype correlation of TRIM8-related neuro-renal syndrome: three cases reports and literature review.
TRIM8-related neuro-renal syndrome (NRS), caused by pathogenic variants of the TRIM8 gene, is characterized by epilepsy, developmental delay (DD) and renal disorders. The severity of the neurological effects as well as the presence of renal disorders is variable among patients. Here, we report three additional patients with clinical features compatible with NRS and summarize the association between the variants' loci and phenotype of TIRM8-related NRS. A retrospective analysis was conducted for three Chinese children with NRS due to TRIM8 variants identified through whole-exome sequencing (WES). Previous reports of patients with TRIM8-related NRS were reviewed systematically. Demographic and clinical data were collected from these patients. Two de novo TRIM8 truncating variants in three NRS patients were identified in our study, including c.1327_c.1328delCCinsTG (p. Arg443*) and c.1375C>T (p.Gln459*). Our three patients all exhibited drug-resistant epilepsy and early-onset DD, and two of whom developed electrical status epilepticus during sleep (ESES). Brain magnetic resonance imaging (MRI) showed periventricular leukomalacia in one patient and normal in the other two. All three patients demonstrated nephrotic range proteinuria (NRP) or nephrotic syndrome (NS) with normal renal function during follow-up. There was a total of 27 patients with TRIM8-related NRS have been identified to date. The most common clinical features are renal diseases (89%), DD (89%), followed by epilepsy (78%). 67% of patients eventually progressed to end-stage renal disease (ESRD). Focal seizure was the most frequent seizure type (57%). 52% of patients presented drug-resistant epilepsy. 64% of patients exhibited non-specific brain MRI abnormalities. Brain atrophy was the most common change (50%). Two patients with TRIM8 variants closer to the N-terminal had neurological diseases without renal damage. Five patients with TRIM8 variants closer to the C-terminal had no severe neurological diseases. Seven patients had Gln459* variant which is the most common variant (7/27, 25.9%). The severity of the renal and neurological damage of the seven patients was variable. This study expands the number of individuals with confirmed NRS due to pathogenic variants in TRIM8. Neurological and renal phenotype with the same variant locus differed in their severity. Further research is needed to explore the relationship between genotype and phenotype of TRIM8 variants.
Clinical and molecular heterogeneity in CDLK5 disorders.
CDKL5 deficiency syndrome is caused by pathogenic variants in the CDKL5 gene, with a variable clinical spectrum ranging from patients with characteristics of autism spectrum disorder to early-onset epilepsy refractory to treatment. Initially, until the gene was discovered, it was considered an atypical form of Rett syndrome. This study aimed to describe the clinical and molecular heterogeneity in CDLK5 disorders among three female patients with CDKL5 pathogenic variants. We reported three unrelated Mexican female patients evaluated for global developmental delay and epilepsy. All three cases were hemizygotes to a CDKL5 pathogenic variant. In one patient, we performed a 306 gene panel associated with epilepsy. In the other two cases, a human genomic microarray was performed. We describe their clinical features electroencephalogram and brain magnetic resonance evaluations. CDKL5 deficiency syndrome represents a challenge for clinicians since the clinical manifestations, electroencephalographic and neuroimaging studies can be non-specific. This syndrome should be suspected in the presence of global developmental delay, autistic behavioral phenotype and epilepsy, associated or not with dysmorphia. Given the similarity between various epileptic encephalopathies, multigene panels including sequencing and duplication/deletion analysis should be requested in which this gene and its possible differential diagnoses are considered, without forgetting the usefulness of genomic techniques in unclear cases. El síndrome por deficiencia de CDKL5 es originado por variantes patogénicas en el gen CDKL5, con un espectro clínico variable que va desde pacientes con características del trastorno del espectro autista hasta epilepsia de inicio temprano y refractaria al tratamiento. Inicialmente fue considerado como una forma atípica de síndrome de Rett. Presentamos tres pacientes no relacionadas, evaluadas por retraso global del desarrollo y epilepsia refractaria. Los tres casos eran hemicigotos a una variante patógena de CDKL5. En una paciente se realizó panel de 306 genes asociados con epilepsia; en las otras dos se realizó microarreglo genómico comparativo. Las características clínicas y los hallazgos en el electroencefalograma y la resonancia magnética cerebral se han descrito clásicamente en el espectro de manifestaciones de este síndrome. El síndrome por deficiencia de CDKL5 representa un reto para los médicos, ya que en muchos casos las manifestaciones clínicas y los estudios electroencefalográficos y de neuroimagen pueden ser inespecíficos. Debe sospecharse este síndrome ante la presencia de retraso global del desarrollo, fenotipo conductual autista y epilepsia, asociado o no con dismorfias. Dada la similitud entre diversas encefalopatías epilépticas, se deben solicitar paneles multigénicos que incluyan la secuenciación y el análisis de duplicación/deleción en los que se contemple este gen y sus posibles diagnósticos diferenciales, aunque sin olvidar la utilidad de las técnicas genómicas en casos poco claros.
A Novel Variant of the CHD2 Gene Associated With Developmental Delay and Myoclonic Epilepsy.
Pathogenic variants in CHD2 have been reported to have a wide range of phenotypic variability in neurodevelopmental disorders, such as early-onset epileptic encephalopathy, developmental delay, and behavior problems. So far, there is no clear correlation between genotypes and phenotypes. This study reports a Chinese patient with a novel heterozygous CHD2 mutation (c.4318C>T, pArg1440*). Her main clinical manifestations include developmental delay, myoclonic epilepsy, and hypothyroidism. Then, we reviewed a total of 144 individuals carrying CHD2 variants with epileptic encephalopathy. In terms of clinical manifestations, these patients are usually described with variable epilepsy phenotypes, including idiopathic photosensitive occipital epilepsy, Dravet syndrome, Jeavons syndrome, Lennox-Gastaut syndrome, juvenile myoclonic epilepsy, and non-specific epileptic encephalopathy. Among them, myoclonic seizures and generalized tonic-clonic seizures are the main seizure types in all patients hosting CHD2 single-nucleotide or indel variants (non-CNVs). At the molecular level, there are 102 types of CHD2 non-CNVs in 126 patients, almost one mutational type corresponding to one person, and there is no difference in the incidence ratio of each position. Furthermore, we summarized that a small proportion of patients inherited CHD2 variants, and not all patients with CHD2 variants had seizures. Importantly, the phenotypes, especially seizures control and fever sensitivity, and genotypes had a relative association. These results enriched the database of CHD2-relative neurodevelopmental disorders and provided a theoretical foundation for researching the relationship between genotypes and phenotypes.
Human COQ4 deficiency: delineating the clinical, metabolic and neuroimaging phenotypes.
Human coenzyme Q4 (COQ4) is essential for coenzyme Q10 (CoQ10) biosynthesis. Pathogenic variants in COQ4 cause childhood-onset neurodegeneration. We aimed to delineate the clinical spectrum and the cellular consequences of COQ4 deficiency. Clinical course and neuroradiological findings in a large cohort of paediatric patients with COQ4 deficiency were analysed. Functional studies in patient-derived cell lines were performed. We characterised 44 individuals from 36 families with COQ4 deficiency (16 newly described). A total of 23 different variants were identified, including four novel variants in COQ4. Correlation analyses of clinical and neuroimaging findings revealed three disease patterns: type 1: early-onset phenotype with neonatal brain anomalies and epileptic encephalopathy; type 2: intermediate phenotype with distinct stroke-like lesions; and type 3: moderate phenotype with non-specific brain pathology and a stable disease course. The functional relevance of COQ4 variants was supported by in vitro studies using patient-derived fibroblast lines. Experiments revealed significantly decreased COQ4 protein levels, reduced levels of cellular CoQ10 and elevated levels of the metabolic intermediate 6-demethoxyubiquinone. Our study describes the heterogeneous clinical presentation of COQ4 deficiency and identifies phenotypic subtypes. Cell-based studies support the pathogenic characteristics of COQ4 variants. Due to the insufficient clinical response to oral CoQ10 supplementation, alternative treatment strategies are warranted.
Phenotypic spectrum of patients with GABRB2 variants: from mild febrile seizures to severe epileptic encephalopathy.
To characterize the different phenotypes of GABRB2-related epilepsy and to establish a genotype-phenotype correlation. We used next-generation sequencing to identify GABRB2 variants in 15 patients. Eleven GABRB2 variants were novel and 12 were de novo. The age at the onset of seizures ranged from 1 day to 26 months. Nine patients had multiple seizure types, including focal seizures, generalized tonic-clonic seizures, myoclonic seizures, epileptic spasms, and atonic seizures. Seizures were fever-sensitive in 13 out of the 15 patients. Eleven patients displayed developmental delay, while 11 had abnormal video electroencephalography. Abnormalities in the brain images included dysplasia of the frontal and temporal cortex, dysplasia of the corpus callosum, and delayed myelination in four patients. One patient was diagnosed with febrile seizures, three with febrile seizures plus, three with Dravet syndrome, three with West syndrome, one with Ohtahara syndrome, three with developmental delays and epilepsy, and one with non-specific early-onset epileptic encephalopathy. The most common phenotypes of patients with GABRB2 variants include early onset of seizure and fever sensitivity. Febrile seizures and febrile seizures plus are new phenotypes of GABRB2 variants. The phenotypic spectrum of GABRB2 variants ranges from mild febrile seizures to severe epileptic encephalopathy. 目的: 总结GABRB2基因相关癫痫的不同表型特点及基因型-表型相关性研究 方法: 采用二代测序技术的方法发现15例患者携带GABRB2基因变异 结果: 15例GABRB2基因变异中11例为新发现的变异,12例为新生变异。癫痫起病年龄范围从生后第1天至26月龄。共有9名患者有多种癫痫发作形式,包括局灶性发作,全面性强直阵挛发作,肌阵挛发作,癫痫性痉挛发作和失张力发作。15例患者中有13例出现癫痫热敏感。11例患者出现发育落后,且有脑电图异常。4例患者出现头颅影像异常,包括额颞区皮质发育不良,胼胝体发育不良,髓鞘化延迟。癫痫诊断包括热性惊厥1例,热性惊厥附加症3例,Dravet综合征3例,婴儿痉挛症3例,大田原综合征1例,发育落后与癫痫3例,不能分类的早发癫痫性脑病1例。 结论: GABRB2基因变异导致的最常见表型包括癫痫起病年龄早和癫痫热敏感。热性惊厥和热性惊厥附加症是GABRB2基因变异的新表型。GABRB2基因变异的表型谱广泛,从表型轻的热性惊厥到严重的癫痫性脑病。.
Publicações recentes
Case report : a novel ASXL3 gene variant in a Sudanese boy.
TBCK-related intellectual disability syndrome: Case study of two patients.
📚 EuropePMCmostrando 8
Variants loci and phenotype correlation of TRIM8-related neuro-renal syndrome: three cases reports and literature review.
Frontiers in neurologyClinical and molecular heterogeneity in CDLK5 disorders.
Boletin medico del Hospital Infantil de MexicoA Novel Variant of the CHD2 Gene Associated With Developmental Delay and Myoclonic Epilepsy.
Frontiers in geneticsHuman COQ4 deficiency: delineating the clinical, metabolic and neuroimaging phenotypes.
Journal of medical geneticsPhenotypic spectrum of patients with GABRB2 variants: from mild febrile seizures to severe epileptic encephalopathy.
Developmental medicine and child neurologyThe value of plasma vitamin B6 profiles in early onset epileptic encephalopathies.
Journal of inherited metabolic diseaseRARS2 mutations cause early onset epileptic encephalopathy without ponto-cerebellar hypoplasia.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyRFT1-congenital disorder of glycosylation (CDG) syndrome: a cause of early-onset severe epilepsy.
Epileptic disorders : international epilepsy journal with videotapeAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Encefalopatia epiléptica de início precoce inespecífica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Encefalopatia epiléptica de início precoce inespecífica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Variants loci and phenotype correlation of TRIM8-related neuro-renal syndrome: three cases reports and literature review.
- Clinical and molecular heterogeneity in CDLK5 disorders.
- A Novel Variant of the CHD2 Gene Associated With Developmental Delay and Myoclonic Epilepsy.
- Human COQ4 deficiency: delineating the clinical, metabolic and neuroimaging phenotypes.
- Phenotypic spectrum of patients with GABRB2 variants: from mild febrile seizures to severe epileptic encephalopathy.
- Case report : a novel ASXL3 gene variant in a Sudanese boy.
- TBCK-related intellectual disability syndrome: Case study of two patients.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:442835(Orphanet)
- MONDO:0018614(MONDO)
- GARD:15028(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56014174(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Encefalopatia epiléptica de início precoce inespecífica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata