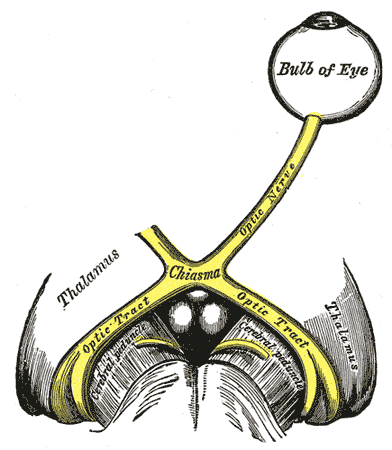
Displasia septo-óptica (DSO), também conhecida como síndrome de de Morsier, é uma síndrome rara de malformação congênita que apresenta uma combinação de subdesenvolvimento do nervo óptico, disfunção da glândula hipófise e ausência do septo pelúcido. Duas ou mais dessas características precisam estar presentes para um diagnóstico clínico — apenas 30% dos pacientes apresentam todas as três. O médico franco-suíço Georges de Morsier foi o primeiro a reconhecer a relação de um septo pelúcido rudimentar ou ausente com hipoplasia dos nervos ópticos e quiasma em 1956.
Introdução
O que você precisa saber de cara
Displasia septo-óptica (DSO), também conhecida como síndrome de de Morsier, é uma síndrome rara de malformação congênita que apresenta uma combinação de subdesenvolvimento do nervo óptico, disfunção da glândula hipófise e ausência do septo pelúcido. Duas ou mais dessas características precisam estar presentes para um diagnóstico clínico — apenas 30% dos pacientes apresentam todas as três. O médico franco-suíço Georges de Morsier foi o primeiro a reconhecer a relação de um septo pelúcido rudimentar ou ausente com hipoplasia dos nervos ópticos e quiasma em 1956.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 6 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 29 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisATP-dependent chaperone part of the 55LCC heterohexameric ATPase complex which is chromatin-associated and promotes replisome proteostasis to maintain replication fork progression and genome stability. Required for replication fork progression, sister chromatid cohesion, and chromosome stability. The ATPase activity is specifically enhanced by replication fork DNA and is coupled to cysteine protease-dependent cleavage of replisome substrates in response to replication fork damage. Uses ATPase ac
CytoplasmCytoplasm, cytoskeleton, spindleNucleus
Deafness, autosomal recessive, 119
A form of non-syndromic deafness characterized by mild to profound sensorineural hearing loss. Sensorineural hearing loss results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information.
Variantes genéticas (ClinVar)
45 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de perda auditiva neurossensorial-quadriplegia espástica-perturbação do desenvolvimento intelectual
Centros de Referência SUS
13 centros habilitados pelo SUS para Síndrome de perda auditiva neurossensorial-quadriplegia espástica-perturbação do desenvolvimento intelectual
Centros para Síndrome de perda auditiva neurossensorial-quadriplegia espástica-perturbação do desenvolvimento intelectual
Detalhes dos centros
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
PRPS1 (p.V42L) Mutation in Arts Syndrome Induces Aberrant Neural Stem Cell Development and Neuronal Senescence-Like Phenotype: Rescue by Nicotinamide Mononucleotide Supplementation.
Arts syndrome is a rare X-linked recessive neurodevelopmental disorder arising from pathogenic variants in PRPS1, which encodes phosphoribosyl pyrophosphate synthetase 1-an enzyme essential for de novo nucleotide biosynthesis. Affected individuals typically exhibit sensorineural hearing loss, intellectual disability, cerebellar ataxia, and recurrent infections. However, despite the severity of these clinical manifestations, therapeutic interventions remain limited, largely due to an incomplete understanding of the cellular pathophysiology underlying the disorder. In this study, we generated patient-specific induced pluripotent stem cells harboring the PRPS1 p.V42L variant and differentiated them into neural stem cells (NSCs) and neurons to elucidate disease mechanisms and explore potential therapeutic strategies. Patient-derived NSCs demonstrated significantly reduced proliferative capacity, aberrant nuclear morphology, and increased neuronal senescence, while mitochondrial integrity and function were largely preserved. Neurons differentiated from these NSCs exhibited impaired neurite outgrowth and reduced branching complexity, indicative of disrupted neurodevelopmental processes. Notably, supplementation with nicotinamide mononucleotide, a precursor of nicotinamide adenine dinucleotide (NAD+), partially ameliorated defects in NSC proliferation, nuclear architecture, and neuronal morphology. Collectively, these findings delineate key cellular mechanisms underlying PRPS1-associated neurodevelopmental pathology and identify NAD+ metabolic augmentation as a promising therapeutic avenue for Arts syndrome and related PRPS1-mediated disorders.
Audiovestibular Dysfunction Related to Long COVID-19 Syndrome: A Systematic Review of Characteristics, Pathophysiology, Diagnosis, and Management.
Long COVID-19 syndrome (or so-called post-COVID-19) is indicated by miscellaneous symptoms, usually starting 3 months from the COVID-19 infection and lasting for at least 2 months, which cannot be explained by an alternative diagnosis. There has been more and more reports addressing the audiovestibular dysfunction related to long COVID-19 syndrome. Emerging evidence suggests that the linkage between audiovestibular dysfunction and long COVID-19 syndrome might rely on (a) direct inner ear system damage related to viral invasion and consequent inflammation, (b) micro thromboembolic events, which might result from the COVID-19-induced autoimmune reaction against endothelial cells, and consequent transient-ischemia and hypoxia of the auditory pathways, (c) the disturbed nerve conduction in vestibulocochlear nerves due to viral invasion, and finally (d) altered auditory cortex function, either imbalanced central gain or neurotransmitter disturbance. However, most of the aforementioned mechanism remained hypothetic and still needed further studies to approve or refute. This systematic review synthesizes current evidence on the characteristics, pathophysiology, diagnostic approaches, and management of audiovestibular dysfunction related to long COVID-19 syndrome. Literature searches across PubMed, Embase, ClinicalKey, Web of Science, and ScienceDirect (up to 15 December 2025) were conducted in accordance with PRISMA guidelines. Through this systematic review, we provided a schematic diagram of the physiopathology of long COVID-19 syndrome-related audiovestibular dysfunction. Further, we summarized the currently available diagnostic tools to explore the audiovestibular function in such patients. The currently available treatment, either pharmacotherapy or nonpharmacotherapy, mainly tackles idiopathic audiovestibular dysfunction but not specifically long COVID-19 syndrome-related audiovestibular dysfunction. Timely recognition and intervention may prevent progression to permanent hearing loss or vestibular disability, improving quality of life. Trial registration: PROSPERO CRD420251265741.
Pre-operative High-Resolution CT and MRI Evaluation in Pediatric Cochlear Implant Candidates: Correlation With Surgical Findings and Outcomes.
Purpose Congenital sensorineural hearing loss (SNHL) is a major childhood disability, and early cochlear implantation offers optimal auditory and language outcomes. High-resolution computed tomography (HRCT) and magnetic resonance imaging (MRI) are essential in evaluating candidacy, identifying inner ear malformations (IEMs), assessing cochlear nerve integrity, and predicting surgical challenges. This study evaluates the role of HRCT and MRI in pediatric cochlear implant candidates and correlates imaging findings with intraoperative events and postoperative outcomes. Methods This retrospective study included 32 children (<7 years) with congenital SNHL who underwent HRCT and MRI of the temporal bones. HRCT assessed mastoid pneumatization, middle ear status, vascular variants, facial recess anatomy, and cochlear morphology. MRI evaluated the membranous labyrinth, cochleovestibular nerve, internal auditory canal (IAC), and brain. Exclusion criteria included lack of consent, age >7 years, metallic implants, pacemakers, or prior cochlear implantation. Surgical records were retrospectively reviewed to assess operative complexity, including duration of surgery, intraoperative challenges, and complications encountered. Results Among 32 patients (mean age 2.9 years), mastoid aeration was normal in 81.25%, and the middle ear cavity was aerated in 90.63%. Sigmoid sinus variants, high-riding jugular bulbs (18.75%), and low-lying dural plates (34.38%) were common. Vestibular aqueduct anomalies were identified in 18.75%. The cochlear aperture was normal in 74.88%, widened in 12.5%, and stenosed in 6.25%. Inner ear malformations were present in 15.63%, including large vestibular aqueduct syndrome (LVAS), incomplete partitions, and cochlear hypoplasia, with cochlear nerve aplasia in two cases. Nineteen patients underwent cochlear implantation; all IEM cases experienced intraoperative perilymphatic gushers, consistent with imaging predictions. All surgeries used the transmastoid facial recess approach with an extended round window technique. Postoperative recovery was uneventful in all cases. Conclusions HRCT and MRI together provide a comprehensive assessment of pediatric cochlear implant candidates, enabling precise identification of IEMs, cochlear nerve anomalies, and surgical risk factors. Imaging reliably predicts intraoperative challenges such as gushers and aids in tailoring surgical approaches. A combined CT-MRI protocol is indispensable for optimizing safety, minimizing complications, and enhancing postoperative outcomes in pediatric cochlear implantation.
The Clinical Details of MYH9-Related Disease and DFNA17 in a Large Japanese Hearing Loss Cohort.
Background/Objectives: MYH9 gene variants cause MYH9-related disease (MYH9-RD), which is also known as Epstein syndrome, Fechtner syndrome, May-Hegglin anomaly, and Sebastian syndrome. MYH9-RD is characterized by sensorineural hearing loss, macrothrombocytopenia, thrombocytopenia, hematuria/proteinuria, glomerulonephritis, cataracts purpura, and mucosal bleeding. In addition, the MYH9 gene is also known to be causative of autosomal dominant non-syndromic hearing loss (DFNA17). MYH9-RD is a relatively rare disorder, and the detailed clinical features and mutational spectra remain unclear. Methods: In this study, we performed next-generation sequencing analysis for 15,684 hearing loss patients and identified MYH9-associated hearing loss patients. Detailed clinical information was collected for these patients and summarized. Results: In this study, we identified 24 patients from 18 families with MYH9-associated hearing loss. We clarified the details of hearing deterioration observed in patients based on collected serial audiogram data. Some cases showed rapid hearing deterioration that worsened by about 50 dB within 5 years. Hearing loss is more likely to progress in patients with myosin head domain variants than in patients with myosin tail domain variants, but hearing loss in each set of patients finally deteriorates to bilateral profound hearing loss. Conclusions: In this study, we were able to clarify the detailed characteristics of MYH9-RD- and DFNA17-related hearing loss in a relatively large number of patients, particularly in some cases that showed rapid and asymmetrical hearing deterioration progressing to bilateral profound hearing loss. Our data will be useful for providing more appropriate treatment and follow-up for MYH9-associated hearing loss.
Novel Mutations in KCNJ10 Gene Associated With SeSAME Syndrome: Rare Disorder With Possible Common Mutation.
Mutations in the KCNJ10 gene cause SeSAME syndrome, an autosomal recessive disorder characterised by seizures, sensorineural deafness, ataxia, intellectual impairment and electrolyte imbalances. KCNJ10 encodes an inwardly rectifying potassium channel Kir4.1, which is essential for preserving potassium ion homeostasis. We assessed three Iranian families with SeSAME syndrome-like symptoms through whole-exome sequencing (WES). Segregation analysis and Sanger sequencing were also used to confirm identified mutations. Additionally, bioinformatic tools were utilised to predict the pathogenicity of the variants. We identified two novel KCNJ10 mutations, c.967 T>C (p.Y323H) and c.352G>A (p.A118T) in three families. While there was no evidence of renal involvement, the probands from these families displayed early-onset seizures, ataxia, developmental delays and hearing abnormalities. Based on the Kir4.1 protein's structural modelling, the stability of the channel is influenced by both mutations. Precisely, p.A118T alters the transmembrane domain that is critical to channel operation, whereas p.Y323H modifies the cytoplasmic C-terminal domain, which may compromise intracellular localisation and regulation. Our findings can expand the spectrum of mutations in the KCNJ10 gene and provide insight into the genotype-phenotype correlation in the SeSAME syndrome.
Publicações recentes
Cochlear implantation in syndromic patients: difficulties and lessons learnt.
An atypical Aymé-Gripp phenotype detected by exome sequencing.
Dominant negative variants in IKZF2 cause ICHAD syndrome, a new disorder characterised by immunodysregulation, craniofacial anomalies, hearing impairment, athelia and developmental delay.
Generation and mutational analysis of a transgenic murine model of the human MAF mutation.
Cogan's syndrome is more than just keratitis: a case-based literature review.
📚 EuropePMCmostrando 199
Pre-operative High-Resolution CT and MRI Evaluation in Pediatric Cochlear Implant Candidates: Correlation With Surgical Findings and Outcomes.
CureusPRPS1 (p.V42L) Mutation in Arts Syndrome Induces Aberrant Neural Stem Cell Development and Neuronal Senescence-Like Phenotype: Rescue by Nicotinamide Mononucleotide Supplementation.
International journal of stem cellsThe Clinical Details of MYH9-Related Disease and DFNA17 in a Large Japanese Hearing Loss Cohort.
GenesNovel Mutations in KCNJ10 Gene Associated With SeSAME Syndrome: Rare Disorder With Possible Common Mutation.
Molecular genetics & genomic medicineAudiovestibular Dysfunction Related to Long COVID-19 Syndrome: A Systematic Review of Characteristics, Pathophysiology, Diagnosis, and Management.
International journal of molecular sciencesA novel CLPP variant in a Pakistani family with Perrault syndrome associated with recurrent fevers.
Clinica chimica acta; international journal of clinical chemistryA pediatric case of diphthamide biosynthesis 1 gene defect presenting with developmental delay, short stature, dysmorphic features, and sparse hair (Loucks-Innes syndrome): a case report.
Journal of medical case reportsExpanding the clinical spectrum: A case report of the first Jordanian presentation of KID syndrome with neurological and skeletal anomalies beyond the classical triad.
MedicineExpanding the Phenotype of Syndromic SLC30A9 -Associated Disease.
American journal of medical genetics. Part AAudiovestibular Dysfunction in Hyper-IgE Syndrome: A Systematic Review of Characteristics, Pathophysiology, Diagnosis, and Management.
International journal of molecular sciencesDe Novo Heterozygous ZFX Frameshift Variant in a Female With an X-Linked Neurodevelopmental Disorder.
American journal of medical genetics. Part AOutcomes of Maintenance Immunotherapy in a Cohort of Patients With Susac Syndrome: A 2-Center Large Case Series.
Neurology. Clinical practiceACTB deletions or single-nucleotide loss-of-function variants: expansion and further delineation of the phenotype and review of the literature.
Journal of medical geneticsOver 30 Years of Neonatal Respiratory Extracorporeal Membrane Oxygenation From a Regional Program.
ASAIO journal (American Society for Artificial Internal Organs : 1992)Bilateral vestibular hypofunction in children.
International journal of pediatric otorhinolaryngology[Clinical phenotype and genotypic analysis of a four-generation Chinese pedigree affected with Stickler syndrome and a literature review].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsCase report of paroxysmal dystonia in a child with KBG syndrome: Expansion of the phenotype and utility of whole exome sequencing.
MedicineExpanding the Clinical Phenotype Associated with the NIN Gene; Report of a Patient with Short Stature, Microcephaly and Hearing Loss.
Archives of Iranian medicineCochlear implantation in children with Bartter syndrome: A case report.
Science progressEvidence of an unprecedented cytoplasmic function of DDX11, the Warsaw breakage syndrome DNA helicase, in regulating autophagy.
AutophagyNovel biallelic NUP107 variants affect the nuclear pore complex and expand the clinical spectrum to include brain malformations.
Journal of medical geneticsTownes-Brocks syndrome: genotype-phenotype correlations of SALL1 variants in our series and the literature.
European journal of human genetics : EJHGHomozygous variants in EIF3K associated with neurodevelopmental delay, microcephaly, and growth retardation.
HGG advancesStudy on Conductive Hearing Loss in Children with Down Syndrome.
Indian journal of otolaryngology and head and neck surgery : official publication of the Association of Otolaryngologists of IndiaNatural history of patients with autosomal dominant WFS1 pathogenic variants associated with sensorineural hearing loss and optic atrophy.
medRxiv : the preprint server for health sciencesBi-allelic variants in MRPL49 cause variable clinical presentations, including sensorineural hearing loss, leukodystrophy, and ovarian insufficiency.
American journal of human geneticsPrevalence and Clinical Characteristics of OTOGL-Associated Hearing Loss Identified in a Cohort of 7065 Japanese Patients with Hearing Loss.
GenesComparison of vHIT deficits with Ramsay Hunt syndrome with dizziness, vestibular neuritis, and idiopathic sudden sensorineural hearing loss with vertigo.
Journal of vestibular research : equilibrium & orientationTwo de novo UBR1 variants in trans as a cause of Johanson-Blizzard syndrome.
Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, CzechoslovakiaGlutamate metabolism disruption in Johanson-Blizzard syndrome: Insights from C. elegans ubr-1 model.
Journal of biosciencesCase Report: The first Korean familial case of BCAP31-related deafness, dystonia, and cerebral hypomyelination.
Frontiers in pediatricsNovel OTOG Variants and Clinical Features of Hearing Loss in a Large Japanese Cohort.
GenesGenetic etiology of Perrault syndrome in Iranian families: first report from Iran and literature review.
Journal of applied geneticsRethinking speech sound disorder (SSD) in non-syndromic cleft lip and palate: The importance of recognizing phonological and language difficulties.
International journal of language & communication disordersSusac's Syndrome: A Tale of Disability Due to Late Recognition.
CureusGenetic Diagnostics and Phenotypic Profiling of a Girl With Autosomal Recessive Intellectual Developmental Disorder and Autism.
CureusClinical Characterization and Prognostic Risk Factors of Susac Syndrome: A Retrospective Multicenter Study.
Neurology(R) neuroimmunology & neuroinflammationJohanson-Blizzard syndrome with cystic dilation of the cochlea and hypoplastic modiolus: a case report.
Pediatric radiologyOcular findings in Baraitser-Winter syndrome with a de novo mutation in the ACTG1 gene: a case report.
BMC ophthalmologyExpanding the genotypic and phenotypic spectrum of EAST/SeSAME syndrome: identification of a novel homozygous mutation (c.194 G > A) in KCNJ10 gene.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyBiallelic variants in MRPL49 cause variable clinical presentations, including sensorineural hearing loss, leukodystrophy, and ovarian insufficiency.
medRxiv : the preprint server for health sciencesIdentification of a novel EYA4 likely pathogenic variant in a Chinese family with postlingual non-syndromic hearing loss and analysis of molecular epidemiology of EYA4 variants.
BMC medical genomicsNeurological manifestations in adult patients with the m.3243A>G variant in mitochondrial DNA.
BMJ neurology openLong-term outcomes in children with riboflavin transporter deficiency and surveillance recommendations.
Developmental medicine and child neurologyInteraction between the TBC1D24 TLDc domain and the KIBRA C2 domain is disrupted by two epilepsy-associated TBC1D24 missense variants.
The Journal of biological chemistryCochlear implantation in syndromic patients: difficulties and lessons learnt.
European archives of oto-rhino-laryngology : official journal of the European Federation of Oto-Rhino-Laryngological Societies (EUFOS) : affiliated with the German Society for Oto-Rhino-Laryngology - Head and Neck SurgeryWhole exome sequencing reveals a dual diagnosis of BCAP31-related syndrome and glutaric aciduria III.
Molecular genetics and metabolism reportsEstablishment of a human induced pluripotent stem cell line (SDQLCHi079-A) from a patient with Johanson-Blizzard syndrome carrying heterozygous mutation in UBR1 gene.
Stem cell researchIdentification of novel variants in BRF1 gene from patient with developmental delay, hearing abnormality, and nervous system anomalies.
International journal of developmental neuroscience : the official journal of the International Society for Developmental NeuroscienceJohanson-Blizzard syndrome caused by novel UBR1 mutation in four Saudi patients.
JPGN reportsMassive pericardial effusion in an infant with Aymé-Gripp syndrome: A case report and review of the literature.
American journal of medical genetics. Part APaternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review.
GenesResolution of severe neurobehavioral difficulties in an individual with Primrose syndrome with sertraline.
American journal of medical genetics. Part ABiallelic variants in Plexin B2 (PLXNB2) cause amelogenesis imperfecta, hearing loss and intellectual disability.
Journal of medical geneticsRetrospective Analysis of the Outcomes of Genetic Testing in Patients Suspected to Have Hereditary Hearing Loss or Deafness.
American journal of audiologyNeurological disease in xeroderma pigmentosum: prospective cohort study of its features and progression.
Brain : a journal of neurologyAn Infant With Primrose Syndrome: A Case Report.
CureusHearing characteristics and otoradiological abnormalities in three patients with novel pathogenic variants of KMT2D-related Kabuki syndrome.
Molecular genetics & genomic medicineExtensive, 3.8 Mb-Sized Deletion of 22q12 in a Patient with Bilateral Schwannoma, Intellectual Disability, Sensorineural Hearing Loss, and Epilepsy.
Molecular syndromologyPaediatric Cogan Syndrome masquerading as IgA vasculitis.
Modern rheumatology case reportsTwo novel cases of biallelic SMPD4 variants with brain structural abnormalities.
NeurogeneticsWhat General Neurologists Should Know about Autoinflammatory Syndromes?
Brain sciencesAn atypical Aymé-Gripp phenotype detected by exome sequencing.
American journal of medical genetics. Part AMultiple Independent Gene Disorders Causing Bardet-Biedl Syndrome, Congenital Hypothyroidism, and Hearing Loss in a Single Indian Patient.
Brain sciencesWhy should multiple dehiscences of the otic capsule be considered before surgically treating patients with superior semicircular canal dehiscence? A radiological monocentric review and a case series.
Frontiers in neurologySusac syndrome: neurological update (clinical features, long-term observational follow-up and management of sixteen patients).
Journal of neurologyBenefit of high-dose oral riboflavin therapy in riboflavin transporter deficiency.
Journal of the peripheral nervous system : JPNSImaging Guide to Inner Ear Malformations: An Illustrative Review.
Current problems in diagnostic radiologyMonoallelic variation in DHX9, the gene encoding the DExH-box helicase DHX9, underlies neurodevelopment disorders and Charcot-Marie-Tooth disease.
American journal of human genetics[Clinical features and genetic analysis of a child with EAST/SeSAME syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsDominant negative variants in IKZF2 cause ICHAD syndrome, a new disorder characterised by immunodysregulation, craniofacial anomalies, hearing impairment, athelia and developmental delay.
Journal of medical geneticsBehavioral Impairment and Amnesia at the Onset of Susac Syndrome.
CureusGeneration and mutational analysis of a transgenic murine model of the human MAF mutation.
American journal of medical genetics. Part AWhole-exome sequencing detected a novel AIFM1 variant in a Han-Chinese family with Cowchock syndrome.
HereditasCogan's syndrome is more than just keratitis: a case-based literature review.
BMC ophthalmologyCation leak through the ATP1A3 pump causes spasticity and intellectual disability.
Brain : a journal of neurologyEvaluation of a Less Invasive Cochlear Implant Surgery in OPA1 Mutations Provoking Deafblindness.
GenesDeep Brain Stimulation for the Management of AIFM1-Related Disabling Tremor: A Case Series.
Pediatric neurologyThe Mitochondrial tRNASer(UCN) Gene: A Novel m.7484A>G Mutation Associated with Mitochondrial Encephalomyopathy and Literature Review.
Life (Basel, Switzerland)A likely pathogenic ACTG1 variant in a child showing partial phenotypic overlap with Baraitser-Winter syndrome.
American journal of medical genetics. Part ALow dose TGF-β1 can improve vohwinkel syndrome by promoting the proliferation of keratinocytes.
Acta histochemica[Epilepsy syndromes associated with hearing loss].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaGriscelli syndrome type 1: a novel pathogenic variant, and review of literature.
Molecular genetics and genomics : MGGAssociation of Genetic Diagnoses for Childhood-Onset Hearing Loss With Cochlear Implant Outcomes.
JAMA otolaryngology-- head & neck surgery[Analysis of a child with Johanson-Blizzard syndrome due to novel compound heterozygous variants of UBR1 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsTwo SOX11 variants cause Coffin-Siris syndrome with a new feature of sensorineural hearing loss.
American journal of medical genetics. Part AObsessive-compulsive symptoms in ACTG1-associated Baraitser-Winter cerebrofrontofacial syndrome.
Journal of neural transmission (Vienna, Austria : 1996)Radiological Findings of Woodhouse-Sakati Syndrome: Cases Reported From Saudi Arabia.
CureusBrain imaging findings in Liberfarb syndrome: hypomyelination and optic nerve and cerebellar atrophy.
Pediatric radiologyBroadening the genotypic and phenotypic spectrum of MAF in three Chinese Han congenital cataracts families.
American journal of medical genetics. Part ABiallelic loss-of-function variants in RABGAP1 cause a novel neurodevelopmental syndrome.
Genetics in medicine : official journal of the American College of Medical GeneticsIdentification and analysis of deletion breakpoints in four Mohr-Tranebjærg syndrome (MTS) patients.
Scientific reportsWhole exome sequencing and transcript analysis discover a novel pathogenic splice site mutation in DCAF17 gene underlying Woodhouse-Sakati syndrome.
Journal of neuroendocrinologyInfluence of Metabolic Syndrome on the Recovery from Idiopathic Sudden Sensorineural Hearing Loss.
International archives of otorhinolaryngologyCochlear nerve deficiency in SOX11-related Coffin-Siris syndrome.
American journal of medical genetics. Part AAn Ethical Analysis of Newborn Congenital Cytomegalovirus Screening.
PediatricsEAST/SeSAME Syndrome and Beyond: The Spectrum of Kir4.1- and Kir5.1-Associated Channelopathies.
Frontiers in physiologyBiallelic PAN2 variants in individuals with a syndromic neurodevelopmental disorder and multiple congenital anomalies.
European journal of human genetics : EJHGEvaluation of Fundus Blood Flow Perfusion in Patients with Diabetic Retinopathy after PPV with Fundus Color Doppler Based on Big Data Mining.
Journal of healthcare engineeringIdentification of a novel microdeletion causative of Nance-Horan syndrome.
Molecular genetics & genomic medicineDe Novo ACTG1 Variant Expands the Phenotype and Genotype of Partial Deafness and Baraitser-Winter Syndrome.
International journal of molecular sciences[Identifications of the novel mutants on MYO7A in a family with non-syndromic hereditary deafness].
Lin chuang er bi yan hou tou jing wai ke za zhi = Journal of clinical otorhinolaryngology head and neck surgeryNovel splicing-site mutation in DCAF17 gene causing Woodhouse-Sakati syndrome in a large consanguineous family.
Journal of clinical laboratory analysisSLC gene mutations and pediatric neurological disorders: diverse clinical phenotypes in a Saudi Arabian population.
Human genetics50 Years Ago in TheJournalofPediatrics: Molecular Diagnostics Determine Underlying Genetic Etiologies for Well-Described Clinical Syndromes.
The Journal of pediatricsRare Clinical Case of Cryopyrin-associated Periodic Syndrome Presented with Ankylosing Spondylitis: A Case Report.
Current rheumatology reviewsSyndromic Deafness Gene ATP6V1B2 Controls Degeneration of Spiral Ganglion Neurons Through Modulating Proton Flux.
Frontiers in cell and developmental biologyA Novel Intronic KMT2D Variant as a Cause of Kabuki Syndrome: A Case Report.
The application of clinical geneticsMaternal mosaicism for a missense variant in the SMS gene that causes Snyder-Robinson syndrome.
Cold Spring Harbor molecular case studiesFurther delineation of the clinical spectrum of White-Sutton syndrome: 12 new individuals and a review of the literature.
European journal of human genetics : EJHGGenetically altered animal models for ATP1A3-related disorders.
Disease models & mechanismsExpanding on the phenotypic spectrum of Woodhouse-Sakati syndrome due to founder pathogenic variant in DCAF17: Report of 58 additional patients from Qatar and literature review.
American journal of medical genetics. Part APhoniatric, Audiological, Orodental and Speech Problems in a Boy with Cardio-Facio-Cutaneous Syndrome Type 3 (CFC 3) Due to a Pathogenic Variant in MAP2K1 - Case Study.
The application of clinical geneticsA Chinese Boy with Mowat-Wilson Syndrome Caused by a 10 bp Deletion in the ZEB2 Gene.
Pharmacogenomics and personalized medicineBenefit of cochlear implantation in a patient with Myhre syndrome.
BMJ case reportsNew insights into Perrault syndrome, a clinically and genetically heterogeneous disorder.
Human geneticsGlial-Specific Deletion of Med12 Results in Rapid Hearing Loss via Degradation of the Stria Vascularis.
The Journal of neuroscience : the official journal of the Society for NeuroscienceAdducted Thumb and Peripheral Polyneuropathy: Diagnostic Supports in Suspecting White-Sutton Syndrome: Case Report and Review of the Literature.
GenesWolf-Hirschhorn Syndrome with Hyperparathyroidism: A Case Report and a Narrative Review of the Literature.
Journal of pediatric geneticsCryopyrin-associated periodic syndrome: a treatable genetic inflammatory condition.
Practical neurologyCorrespondence on "DOORS syndrome and a recurrent truncating ATP6V1B2 variant" by Beauregard-Lacroix et al.
Genetics in medicine : official journal of the American College of Medical GeneticsKallmann syndrome in a patient with Weiss-Kruszka syndrome and a de novo deletion in 9q31.2.
European journal of endocrinologyEIF3F-related neurodevelopmental disorder: refining the phenotypic and expanding the molecular spectrum.
Orphanet journal of rare diseasesA Novel Synergistic Association of Variants in PTRH2 and KIF1A Relates to a Syndrome of Hereditary Axonopathy, Outer Hair Cell Dysfunction, Intellectual Disability, Pancreatic Lipomatosis, Diabetes, Cerebellar Atrophy, and Vertebral Artery Hypoplasia.
CureusGjb3 Gene Mutations in Non-Syndromic Hearing Loss of Bloch, Kurd, and Turkmen Ethnicities in Iran.
Iranian journal of public healthNew Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform.
International journal of molecular sciencesAuditory phenotype of Smith-Lemli-Opitz syndrome.
American journal of medical genetics. Part AAyme gripp syndrome in an Indian patient.
American journal of medical genetics. Part ADevelopmental performance between pediatric cochlear implantation candidates with and without large vestibular aqueduct syndrome.
Acta oto-laryngologicaDiagnosis and patterns of hearing loss in children with severe developmental delay.
American journal of otolaryngologyBiallelic deletion in a minimal CAPN15 intron in siblings with a recognizable syndrome of congenital malformations and developmental delay.
Clinical geneticsPIGF deficiency causes a phenotype overlapping with DOORS syndrome.
Human geneticsColoboma may be a shared feature in a spectrum of disorders caused by mutations in the WDR37-PACS1-PACS2 axis.
American journal of medical genetics. Part AUBR7 functions with UBR5 in the Notch signaling pathway and is involved in a neurodevelopmental syndrome with epilepsy, ptosis, and hypothyroidism.
American journal of human geneticsAudiological and otologic manifestations of glutaric aciduria type I.
Orphanet journal of rare diseasesBasedow-Graves' disease in a pediatric patient with Sticlker syndrome, a new endocrine finding to improve personalized treatment.
Italian journal of pediatricsDYRK1A pathogenic variants in two patients with syndromic intellectual disability and a review of the literature.
Molecular genetics & genomic medicineDescription of a peculiar alternating ictal electroclinical pattern in a young boy with a novel SPATA5 mutation.
Epileptic disorders : international epilepsy journal with videotapeChudley-McCullough Syndrome: A Recognizable Clinical Entity Characterized by Deafness and Typical Brain Malformations.
Journal of child neurologyDeafness, onychodystrophy, osteodystrophy, mental retardation, and seizures (DOORS) syndrome: a new case report from Indonesia and review of the literature.
European journal of dermatology : EJDCoffin-Siris Syndrome 4-Related Spectrum in a Young Woman Caused by a Heterozygous SMARCA4 Deletion Detected by High-Resolution aCGH.
Molecular syndromologyExpanding the phenotype of cerebellar-facial-dental syndrome: Two siblings with a novel variant in BRF1.
American journal of medical genetics. Part ASphenoid sinus agenesis and sella turcica hypoplasia: very rare cases of two brothers with Hamamy syndrome.
Surgical and radiologic anatomy : SRAReducing the Emergency Department Revolving Door Syndrome for the Poor, Uninsured, and Chronically Ill Patient in Los Angeles: Process Improvement Recommendations from a County Health Program Evaluation.
Population health managementImpact of breast milk-acquired cytomegalovirus infection in premature infants: Pathogenesis, prevention, and clinical consequences?
Reviews in medical virologySLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect.
Brain : a journal of neurologyMild form of Zellweger Spectrum Disorders (ZSD) due to variants in PEX1: Detailed clinical investigation in a 9-years-old female.
Molecular genetics and metabolism reports[Influence of capsular pseudoexfoliation syndrome on hearing loss].
Revista de la Facultad de Ciencias Medicas (Cordoba, Argentina)An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs.
Human molecular geneticsClassification of aplasia cutis congenita: a 25-year review of cases presenting to a tertiary paediatric dermatology department.
Clinical and experimental dermatologyUnique skeletal manifestations in patients with Primrose syndrome.
European journal of medical geneticsEtiology of Childhood Bilateral Sensorineural Hearing Loss in Shandong Province, China.
American journal of audiologyCongenital chloride diarrhea and Pendred syndrome: case report of siblings with two rare recessive disorders of SLC26 family genes.
BMC medical geneticsExome sequencing identifies PEX6 mutations in three cases diagnosed with Retinitis Pigmentosa and hearing impairment.
Molecular visionAnalysis of Etiologic Factors in Pediatric Rhegmatogenous Retinal Detachment With Genetic Testing.
American journal of ophthalmologyBiallelic TANGO1 mutations cause a novel syndromal disease due to hampered cellular collagen secretion.
eLifeNovel mutations in the KCNJ10 gene associated to a distinctive ataxia, sensorineural hearing loss and spasticity clinical phenotype.
Neurogenetics[Susac syndrome - an unusual syndrome which can be mistaken for multiple sclerosis].
LakartidningenSevere forms of Johanson-Blizzard syndrome caused by two novel compound heterozygous variants in UBR1: Clinical manifestations, imaging findings and molecular genetics.
Pancreatology : official journal of the International Association of Pancreatology (IAP) ... [et al.]Novel KIAA1033/WASHC4 mutations in three patients with syndromic intellectual disability and a review of the literature.
American journal of medical genetics. Part AAssociated syndromes in patients with Pierre Robin Sequence.
International journal of pediatric otorhinolaryngologyCochlear Implantation Outcomes in Children with Agenesis of the Corpus Callosum: A Retrospective Study and A Review of the Literature.
The journal of international advanced otologyAutosomal dominant inheritance in a recently described ZMIZ1-related neurodevelopmental disorder: Case report of siblings and an affected parent.
American journal of medical genetics. Part AHomozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
American journal of medical genetics. Part AHomozygous Mutation in TWNK Cases Ataxia, Sensorineural Hearing Loss and Optic Nerve Atrophy.
Archives of Iranian medicineA prenatally diagnosed case of Donnai-Barrow syndrome: Highlighting the importance of whole exome sequencing in cases of consanguinity.
American journal of medical genetics. Part ADeficiencies in vesicular transport mediated by TRAPPC4 are associated with severe syndromic intellectual disability.
Brain : a journal of neurologyPatterns of neurological manifestations in Woodhouse-Sakati Syndrome.
Parkinsonism & related disordersPancreatic Malnutrition in Children.
Pediatric annalsComplete oculocerebrorenal phenotype of Lowe syndrome in a female patient with half reduction of inositol polyphosphate 5-phosphatase.
CEN case reportsFunction of hTim8a in complex IV assembly in neuronal cells provides insight into pathomechanism underlying Mohr-Tranebjærg syndrome.
eLifeIdentification and functional characterization of two novel mutations in KCNJ10 and PI4KB in SeSAME syndrome without electrolyte imbalance.
Human genomicsSkeletal abnormalities are common features in Aymé-Gripp syndrome.
Clinical geneticsSusac Syndrome: A Rare Case Report.
Mymensingh medical journal : MMJThe Prevalence and Clinical Characteristics of TECTA-Associated Autosomal Dominant Hearing Loss.
Genes[One case report of Mohr-Tranebjærg syndrome].
Lin chuang er bi yan hou tou jing wai ke za zhi = Journal of clinical otorhinolaryngology head and neck surgery[One novel pathologic variation in KMT2D cause Kabuki syndrome with hearing loss as the main phenotype and related research on types of deafness].
Lin chuang er bi yan hou tou jing wai ke za zhi = Journal of clinical otorhinolaryngology head and neck surgeryNovel de novo interstitial deletion in 2q36.1q36.3 causes syndromic hearing loss and further delineation of the 2q36 deletion syndrome.
Acta oto-laryngologicaMaternally inherited MAF variant associated with variable expression of Aymé-Gripp syndrome.
American journal of medical genetics. Part AWoodhouse-Sakati Syndrome: First report of a Portuguese case.
American journal of medical genetics. Part AStudy of carrier frequency of Warsaw breakage syndrome in the Ashkenazi Jewish population and presentation of two cases.
American journal of medical genetics. Part ANovel homozygous TSFM pathogenic variant associated with encephalocardiomyopathy with sensorineural hearing loss and peculiar neuroradiologic findings.
NeurogeneticsThe Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in thePISD gene.
Genetics in medicine : official journal of the American College of Medical GeneticsMutations in PIGB Cause an Inherited GPI Biosynthesis Defect with an Axonal Neuropathy and Metabolic Abnormality in Severe Cases.
American journal of human geneticsA case of Coffin-Siris syndrome with severe congenital heart disease and a novel SMARCA4 variant.
Cold Spring Harbor molecular case studiesPOGZ-related epilepsy: Case report and review of the literature.
American journal of medical genetics. Part AGenetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies.
Journal of inherited metabolic diseaseEpilepsy in patients with EAST syndrome caused by mutation in the KCNJ10.
Brain & developmentTwo further patients with Warsaw breakage syndrome. Is a mild phenotype possible?
Molecular genetics & genomic medicineNovel compound heterozygous CDH23 variants in a patient with Usher syndrome type I.
Human genome variationSevere cryopyrin-associated periodic syndrome first characterized by early childhood-onset sensorineural hearing loss - Case report and literature review.
International journal of pediatric otorhinolaryngologyXq22.3q23 microdeletion harboring TMEM164 and AMMECR1 genes: Two case reports confirming a recognizable phenotype with short stature, midface hypoplasia, intellectual delay, and elliptocytosis.
American journal of medical genetics. Part AA homozygous splice site ROBO1 mutation in a patient with a novel syndrome with combined pituitary hormone deficiency.
Journal of human geneticsRefining the Primrose syndrome phenotype: A study of five patients with ZBTB20 de novo variants and a review of the literature.
American journal of medical genetics. Part ADOOR syndrome: A case report and its embryological basis.
International journal of pediatric otorhinolaryngologyNovel VPS33B mutation in a patient with autosomal recessive keratoderma-ichthyosis-deafness syndrome.
American journal of medical genetics. Part ACornelia De Lange Syndrome and Cochlear Implantation.
Iranian journal of otorhinolaryngologyDeletion 7q21.2-q22.1 in a case with split hand-split foot malformation, sensorineural hearing loss and intellectual disability: Phenotype subtypes and the correlation with genotypes.
European journal of medical geneticsTissue Specificity in Trisomy 22 Mosaicism: A Tale of Caution for Interpretation of Chromosomal Microarray Results.
Journal of the Association of Genetic TechnologistsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de perda auditiva neurossensorial-quadriplegia espástica-perturbação do desenvolvimento intelectual.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de perda auditiva neurossensorial-quadriplegia espástica-perturbação do desenvolvimento intelectual
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- PRPS1 (p.V42L) Mutation in Arts Syndrome Induces Aberrant Neural Stem Cell Development and Neuronal Senescence-Like Phenotype: Rescue by Nicotinamide Mononucleotide Supplementation.
- Audiovestibular Dysfunction Related to Long COVID-19 Syndrome: A Systematic Review of Characteristics, Pathophysiology, Diagnosis, and Management.
- Pre-operative High-Resolution CT and MRI Evaluation in Pediatric Cochlear Implant Candidates: Correlation With Surgical Findings and Outcomes.
- The Clinical Details of MYH9-Related Disease and DFNA17 in a Large Japanese Hearing Loss Cohort.
- Novel Mutations in KCNJ10 Gene Associated With SeSAME Syndrome: Rare Disorder With Possible Common Mutation.
- Cochlear implantation in syndromic patients: difficulties and lessons learnt.
- An atypical Aymé-Gripp phenotype detected by exome sequencing.
- Dominant negative variants in IKZF2 cause ICHAD syndrome, a new disorder characterised by immunodysregulation, craniofacial anomalies, hearing impairment, athelia and developmental delay.
- Generation and mutational analysis of a transgenic murine model of the human MAF mutation.
- Cogan's syndrome is more than just keratitis: a case-based literature review.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:659975(Orphanet)
- OMIM OMIM:619616(OMIM)
- MONDO:0859206(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de perda auditiva neurossensorial-quadriplegia espástica-perturbação do desenvolvimento intelectual
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS