A miopatia causada pelo excesso das proteínas calsequestrina e SERCA1 se caracteriza por uma fraqueza muscular leve ou por níveis elevados da enzima creatina quinase no sangue, sem que a pessoa sinta outros sintomas.
Introdução
O que você precisa saber de cara
A miopatia causada pelo excesso das proteínas calsequestrina e SERCA1 se caracteriza por uma fraqueza muscular leve ou por níveis elevados da enzima creatina quinase no sangue, sem que a pessoa sinta outros sintomas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 9 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Unknown.
Curadoria gene-doença
fontes oficiaisCalsequestrin is a high-capacity, moderate affinity, calcium-binding protein and thus acts as an internal calcium store in muscle (PubMed:28895244). Calcium ions are bound by clusters of acidic residues at the protein surface, often at the interface between subunits. Can bind around 80 Ca(2+) ions (PubMed:28895244). Regulates the release of lumenal Ca(2+) via the calcium release channel RYR1; this plays an important role in triggering muscle contraction. Negatively regulates store-operated Ca(2+
Endoplasmic reticulumSarcoplasmic reticulumSarcoplasmic reticulum lumenSarcoplasmic reticulum membraneMitochondrion matrix
Myopathy, vacuolar, with CASQ1 aggregates
An autosomal dominant mild muscle disorder characterized by adult onset of muscle cramping and weakness as well as increased levels of serum creatine kinase. The disorder is not progressive, and some patients may be asymptomatic.
Variantes genéticas (ClinVar)
49 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Miopatia por sobrecarga das proteínas calsequestrina e SERCA1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Vacuolar myopathy caused by CASQ1 p.Asp244His: pathogenic evidence from two unrelated Chinese families.
Calsequestrin-1 (CASQ1)-related myopathy is a rare skeletal muscle disorder caused by mutations in CASQ1 gene, which encodes a major calcium-buffering protein of the sarcoplasmic reticulum (SR). It is characterized histopathologically by tubular aggregates or optically empty vacuoles, predominantly affecting type II muscle fibers. In this study, we report two unrelated Chinese patients presenting with late-onset, slowly progressive muscle weakness, fatigue, and myalgia. Both had mildly to moderately elevated serum creatine kinase levels. Muscle biopsies revealed typical optically empty vacuoles primarily in type II fibers. Whole-exome sequencing identified an identical heterozygous CASQ1 variant, c.730G > C (p.Asp244His), located at a highly conserved residue. In vitro expression of the mutant CASQ1 in HeLa cells confirmed its aggregation tendency, suggesting impaired protein folding or calcium handling. Immunofluorescence revealed abnormal aggregation of CASQ1 protein around the edge of vacuoles, co-localized with SQSTM1/p62, and the endoplasmic reticulum (ER) stress marker PERK. Our findings support the pathogenic role of the p.Asp244His variant and provide further insights into CASQ1-related myopathy in Asian populations.
Pathological mechanisms of vacuolar aggregate myopathy arising from a Casq1 mutation.
Mice with a mutation (D244G, DG) in calsequestrin 1 (CASQ1), analogous to a human mutation in CASQ1 associated with a delayed onset human myopathy (vacuolar aggregate myopathy), display a progressive myopathy characterized by decreased activity, decreased ability of fast twitch muscles to generate force and low body weight after one year of age. The DG mutation causes CASQ1 to partially dissociate from the junctional sarcoplasmic reticulum (SR) and accumulate in the endoplasmic reticulum (ER). Decreased junctional CASQ1 reduces SR Ca2+ release. Muscles from older DG mice display ER stress, ER expansion, increased mTOR signaling, inadequate clearance of aggregated proteins by the proteasomes, and elevation of protein aggregates and lysosomes. This study suggests that the myopathy associated with the D244G mutation in CASQ1 is driven by CASQ1 mislocalization, reduced SR Ca2+ release, CASQ1 misfolding/aggregation and ER stress. The subsequent maladaptive increase in protein synthesis and decreased protein aggregate clearance are likely to contribute to disease progression.
The clinical spectrum of CASQ1-related myopathy.
To identify and characterize patients with calsequestrin 1 (CASQ1)-related myopathy. Patients selected according to histopathologic features underwent CASQ1 genetic screening. CASQ1-mutated patients were clinically evaluated and underwent muscle MRI. Vacuole morphology and vacuolated fiber type were characterized. Twenty-two CASQ1-mutated patients (12 families) were identified, 21 sharing the previously described founder mutation (p.Asp244Gly) and 1 with the p.Gly103Asp mutation. Patients usually presented in the sixth decade with exercise intolerance and myalgias and later developed mild to moderate, slowly progressive proximal weakness with quadriceps atrophy and scapular winging. Muscle MRI (n = 11) showed a recurrent fibrofatty substitution pattern. Three patients presented subclinical cardiac abnormalities. Muscle histopathology in patients with p.Asp244Gly showed vacuoles in type II fibers appearing empty in hematoxylin-eosin, Gomori, and nicotinamide adenine dinucleotide (NADH) tetrazolium reductase stains but strongly positive for sarcoplasmic reticulum proteins. The muscle histopathology of p.Gly103Asp mutation was different, showing also NADH-positive accumulation consistent with tubular aggregates. We report the clinical and molecular details of the largest cohort of CASQ1-mutated patients. A possible heart involvement is presented, further expanding the phenotype of the disease. One mutation is common due to a founder effect, but other mutations are possible. Because of a paucity of symptoms, it is likely that CASQ1 mutations may remain undiagnosed if a muscle biopsy is not performed.
Publicações recentes
A Homozygous ATP2A2 Variant Alters Sarcoendoplasmic Reticulum Ca(2+)-ATPase 2 Function in Skeletal Muscle and Causes a Novel Vacuolar Myopathy.
Pathological mechanisms of vacuolar aggregate myopathy arising from a Casq1 mutation.
The clinical spectrum of CASQ1-related myopathy.
A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates.
Myopathy in Marinesco-Sjögren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology.
📚 EuropePMCmostrando 3
Vacuolar myopathy caused by CASQ1 p.Asp244His: pathogenic evidence from two unrelated Chinese families.
Journal of human geneticsPathological mechanisms of vacuolar aggregate myopathy arising from a Casq1 mutation.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyThe clinical spectrum of CASQ1-related myopathy.
NeurologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Miopatia por sobrecarga das proteínas calsequestrina e SERCA1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Miopatia por sobrecarga das proteínas calsequestrina e SERCA1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Vacuolar myopathy caused by CASQ1 p.Asp244His: pathogenic evidence from two unrelated Chinese families.
- Pathological mechanisms of vacuolar aggregate myopathy arising from a Casq1 mutation.FASEB journal : official publication of the Federation of American Societies for Experimental Biology· 2021· PMID 33786938mais citado
- The clinical spectrum of CASQ1-related myopathy.
- A Homozygous ATP2A2 Variant Alters Sarcoendoplasmic Reticulum Ca(2+)-ATPase 2 Function in Skeletal Muscle and Causes a Novel Vacuolar Myopathy.
- A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates.
- Myopathy in Marinesco-Sjögren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:88635(Orphanet)
- OMIM OMIM:616231(OMIM)
- MONDO:0014546(MONDO)
- GARD:16770(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55784876(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
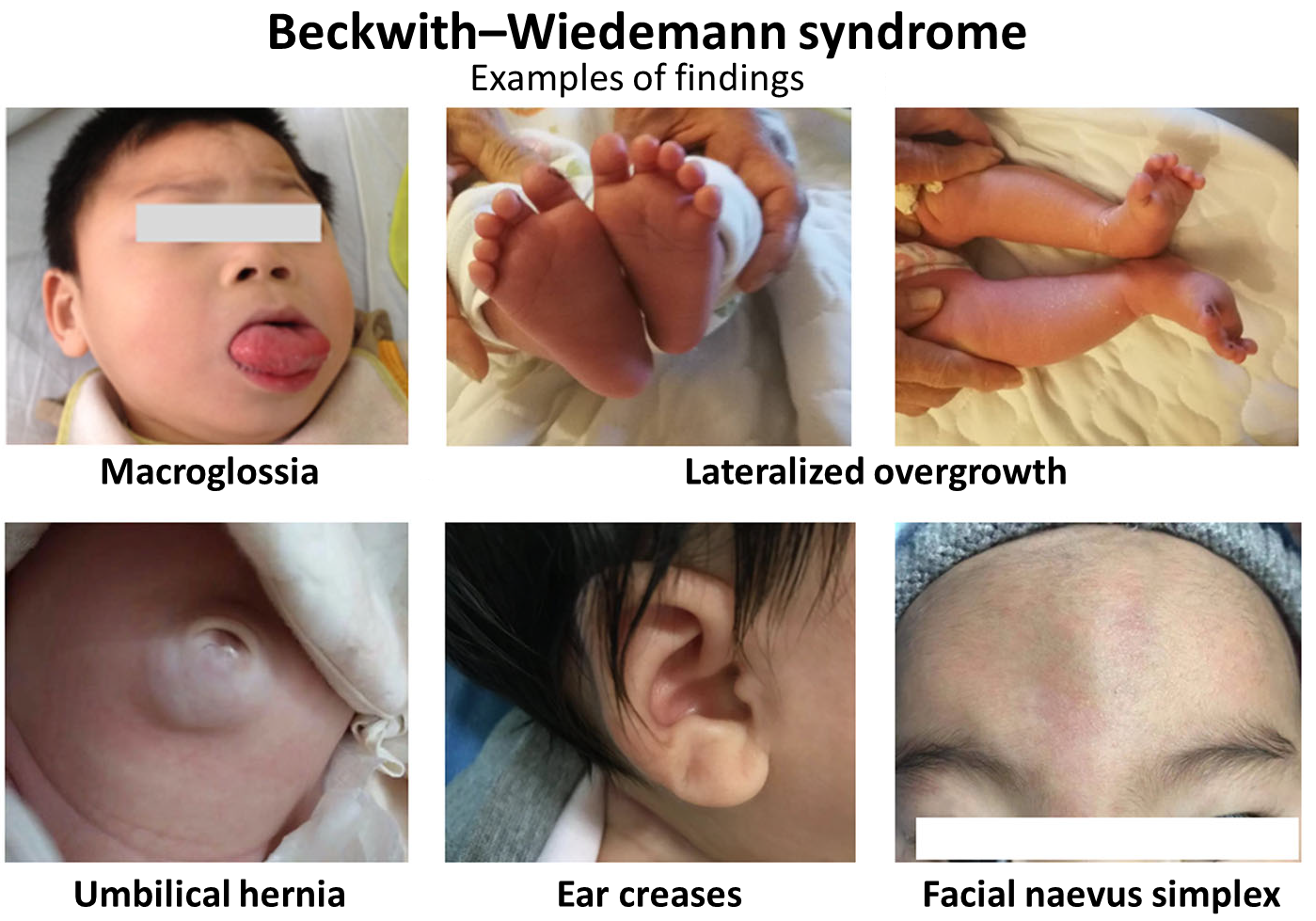
Miopatia por sobrecarga das proteínas calsequestrina e SERCA1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata