A síndrome da coluna rígida (RSS) é uma distrofia muscular congênita de início lento e progressiva, caracterizada por contraturas dos músculos extensores da coluna vertebral associadas à postura anormal (limitação da flexão do pescoço e do tronco), escoliose progressiva da coluna, fraqueza muscular cervico-axial precocemente acentuada com força e função relativamente preservadas das extremidades e insuficiência respiratória progressiva.
Introdução
O que você precisa saber de cara
A síndrome da coluna rígida (RSS) é uma distrofia muscular congênita de início lento e progressiva, caracterizada por contraturas dos músculos extensores da coluna vertebral associadas à postura anormal (limitação da flexão do pescoço e do tronco), escoliose progressiva da coluna, fraqueza muscular cervico-axial precocemente acentuada com força e função relativamente preservadas das extremidades e insuficiência respiratória progressiva.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 15 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisPlays an important role in cell protection against oxidative stress and in the regulation of redox-related calcium homeostasis. Regulates the calcium level of the ER by protecting the calcium pump ATP2A2 against the oxidoreductase ERO1A-mediated oxidative damage. Within the ER, ERO1A activity increases the concentration of H(2)O(2), which attacks the luminal thiols in ATP2A2 and thus leads to cysteinyl sulfenic acid formation (-SOH) and SEPN1 reduces the SOH back to free thiol (-SH), thus restor
Endoplasmic reticulum membrane
Congenital myopathy 3 with rigid spine
An autosomal recessive, slowly progressive muscular disorder apparent from birth or early childhood and characterized by hypotonia, proximal muscle weakness, poor axial muscle strength, scoliosis and neck weakness, and a variable degree of spinal rigidity. Most patients remain ambulatory. Early ventilatory insufficiency may lead to death by respiratory failure. Additional features may include facial muscle weakness, amyotrophy, joint contractures, distal hyperlaxity, pulmonary hypertension with secondary cardiac dysfunction, and insulin resistance in patients with a low BMI. Skeletal muscle biopsy typically shows multiminicores and other abnormal non-specific myopathic findings.
Actins are highly conserved proteins that are involved in various types of cell motility and are ubiquitously expressed in all eukaryotic cells
Cytoplasm, cytoskeleton
Congenital myopathy 2A, typical, autosomal dominant
A muscular disorder characterized by generalized muscle weakness, delayed motor milestones, hypotonia, and muscle fiber abnormalities on histologic examination. Histologic findings include abnormal thread- or rod-like structures (nemaline rods), intranuclear rods, clumped filaments, cores, or fiber-type disproportion. The spectrum of clinical phenotypes ranges from severe neonatal presentations to onset of a milder disorder in childhood.
Variantes genéticas (ClinVar)
493 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 7 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome da coluna rígida
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Novel missense variants in CFL2 affect F-actin depolymerisation and expand the disease spectrum of CFL2-related myopathy.
Cofilin-2, encoded by CFL2, is an actin-binding protein essential for regulating actin filament dynamics in skeletal muscle. Biallelic variants in CFL2 are associated with an ultra-rare, early-onset myopathy typically presenting as nemaline myopathy. Only 10 patients have been described to date from five unrelated families. Here, we describe two new cases from two unrelated families. The first proband presented clinically with rigid spine syndrome and a biopsy keeping with nemaline myopathy. The second proband presented with a relatively mild congenital myopathy which became rapidly progressive in the fourth decade, the muscle biopsy showed cytoplasmic bodies, internal nuclei and ringbinden. Exome and genome sequencing revealed three novel biallelic missense variants in CFL2, a homozygous c.115 T > G; p.(Cys39Gly) in the proband of Family 1, and bi-allelic heterozygous c.256G > A:(p.Asp86Asn), and c.283A > G (p.Lys95Glu) variants in the proband of Family 2. We characterised the effects of these substitutions using an in vitro F-actin depolymerisation assay and showed all three were associated with significantly reduced filamentous actin depolymerisation rates compared to the wildtype. Taken together, our findings are highly suggestive of a CFL2-related disease in these patients. Since CFL2-related myopathies are ultrarare, the application of ACMG/AMP guidelines and the diagnostic reportability of CFL2 variants identified in patients remains a challenge. The actin depolymerisation assay may be useful to elucidate the impact and pathogenicity of additional CFL2 variants and has the potential to guide variant classification in future.
New Clinical Phenotype in a Child Presenting With an FHL1 Mutation.
There is a range of phenotypes associated with pathogenic variants in the FHL1 gene, including X-linked dominant scapuloperoneal myopathy, X-linked myopathy with postural muscle atrophy, reducing body myopathy, Emery-Dreifuss muscular dystrophy, rigid-spine syndrome, and hypertrophic cardiomyopathy. This gene encodes the four-and-a-half LIM domain protein 1 which is highly expressed in skeletal and cardiac muscle. The function of this protein includes influencing cellular architecture, myoblast differentiation, mechanotransduction, and skeletal muscle fiber size. We report a case of a 6-year-old boy with a novel FHL1 gene mutation who presented to the neuromuscular clinic for evaluation of stiffness, joint contractures, and mild proximal weakness. Symptoms first noted in the newborn period have been slowly progressive. The child's presentation has not been described before and represents a new clinical phenotype within the spectrum of FHL1-related disorders.
HMGCS1 variants cause rigid spine syndrome amenable to mevalonic acid treatment in an animal model.
Rigid spine syndrome is a rare childhood-onset myopathy characterized by slowly progressive or non-progressive scoliosis, neck and spine contractures, hypotonia and respiratory insufficiency. Biallelic variants in SELENON account for most cases of rigid spine syndrome, however, the underlying genetic cause in some patients remains unexplained. We used exome and genome sequencing to investigate the genetic basis of rigid spine syndrome in patients without a genetic diagnosis. In five patients from four unrelated families, we identified biallelic variants in HMGCS1 (3-hydroxy-3-methylglutaryl-coenzyme A synthase). These included six missense variants and one frameshift variant distributed throughout HMGCS1. All patients presented with spinal rigidity primarily affecting the cervical and dorso-lumbar regions, scoliosis and respiratory insufficiency. Creatine kinase levels were variably elevated. The clinical course worsened with intercurrent disease or certain drugs in some patients; one patient died from respiratory failure following infection. Muscle biopsies revealed irregularities in oxidative enzyme staining with occasional internal nuclei and rimmed vacuoles. HMGCS1 encodes a critical enzyme of the mevalonate pathway and has not yet been associated with disease. Notably, biallelic hypomorphic variants in downstream enzymes including HMGCR and GGPS1 are associated with muscular dystrophy resembling our cohort's presentation. Analyses of recombinant human HMGCS1 protein and four variants (p.S447P, p.Q29L, p.M70T, p.C268S) showed that all mutants maintained their dimerization state. Three of the four mutants exhibited reduced thermal stability, and two mutants showed subtle changes in enzymatic activity compared to the wildtype. Hmgcs1 mutant zebrafish displayed severe early defects, including immobility at 2 days and death by Day 3 post-fertilisation and were rescued by HMGCS1 mRNA. We demonstrate that the four variants tested (S447P, Q29L, M70T and C268S) have reduced function compared to wild-type HMGCS1 in zebrafish rescue assays. Additionally, we demonstrate the potential for mevalonic acid supplementation to reduce phenotypic severity in mutant zebrafish. Overall, our analyses suggest that these missense variants in HMGCS1 act through a hypomorphic mechanism. Here, we report an additional component of the mevalonate pathway associated with disease and suggest biallelic variants in HMGCS1 should be considered in patients presenting with an unresolved rigid spine myopathy phenotype. Additionally, we highlight mevalonoic acid supplementation as a potential treatment for patients with HMGCS1-related disease.
SEPN1 Related Myopathy Presenting as Chronic Respiratory Insufficiency.
The FHL1 myopathy spectrum revisited: a literature review and report of two new patients.
Mutations in the FHL1 gene have been associated with a diverse spectrum of X-linked diseases affecting skeletal and cardiac muscle. Six clinically distinct human myopathies can be recognized, including reducing body myopathy (RBM), X-linked dominant scapuloperoneal myopathy (SPM), X-linked myopathy with postural muscle atrophy (XMPMA), rigid spine syndrome (RSS), hypertrophic cardiomyopathy (HCM) and type 6 Emery- Dreifuss muscular dystrophy (EDMD). The core features of all described FHL1opathies are mostly scapuloperoneal muscle weakness, rigid spine, cardiac involvement, and cytoplasmic bodies in the muscle biopsy. We systematically reviewed the medical literature between the years 2000 and 2024 regarding the phenotype and genotype description of FHL1-associated myopathies. Here, we report two novel patients presenting with an X-linked myopathy with postural muscle atrophy (XMPMA) caused by the c.672 C > G FHL1 gene mutation. When encountering these features in a patient, one may consider screening for an FHL1 mutation. The course ranges from a severe fatal course with early onset to very mild features with late onset. Once a dystrophinopathy has been excluded, increased CK values in male subjects with possible X-linked inheritance should always trigger FHL1 gene screening.
Publicações recentes
Novel missense variants in CFL2 affect F-actin depolymerisation and expand the disease spectrum of CFL2-related myopathy.
New Clinical Phenotype in a Child Presenting With an FHL1 Mutation.
The FHL1 myopathy spectrum revisited: a literature review and report of two new patients.
HMGCS1 variants cause rigid spine syndrome amenable to mevalonic acid treatment in an animal model.
Genotype-phenotype correlation in recessive DNAJB4 myopathy.
📚 EuropePMC90 artigos no totalmostrando 29
Novel missense variants in CFL2 affect F-actin depolymerisation and expand the disease spectrum of CFL2-related myopathy.
Human molecular geneticsNew Clinical Phenotype in a Child Presenting With an FHL1 Mutation.
Journal of child neurologyThe FHL1 myopathy spectrum revisited: a literature review and report of two new patients.
Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of MyologyHMGCS1 variants cause rigid spine syndrome amenable to mevalonic acid treatment in an animal model.
Brain : a journal of neurologyGenotype-phenotype correlation in recessive DNAJB4 myopathy.
Research squareGenotype‒phenotype correlation in recessive DNAJB4 myopathy.
Acta neuropathologica communicationsMiyoshi myopathy associated with spine rigidity and multiple contractures: a case report.
BMC musculoskeletal disordersSELENON-Related Myopathy Across the Life Span, a Cross-Sectional Study for Preparing Trial Readiness.
Journal of neuromuscular diseasesSEPN1 Related Myopathy Presenting as Chronic Respiratory Insufficiency.
Indian journal of pediatricsCongenital myopathy presenting as recurrent pneumonia with lung collapse and pulmonary artery hypertension.
BMJ case reportsCardiac involvement in two rare neuromuscular diseases: LAMA2-related muscular dystrophy and SELENON-related myopathy.
Neuromuscular disorders : NMDLate Onset Pompe Disease with Novel Mutations and Atypical Phenotypes.
Journal of neuromuscular diseasesRigid Spine Muscular Dystrophy Type 1 Presenting with Neck Tilt.
The Journal of pediatricsPathogenic Mutations and Putative Phenotype-Affecting Variants in Polish Myofibrillar Myopathy Patients.
Journal of clinical medicineThe first report of two homozygous sequence variants in FKRP and SELENON genes associated with syndromic congenital muscular dystrophy in Iran: Further expansion of the clinical phenotypes.
The journal of gene medicineFHL1-related myopathy may not be classified by reducing bodies in muscle biopsy.
Neuromuscular disorders : NMDScreening for late-onset Pompe disease in Poland.
Acta neurologica ScandinavicaA novel mutation in SEPN1 causing rigid spine muscular dystrophy 1: a Case report.
BMC medical geneticsA child diagnosed with rigid spine syndrome complicated by ventilatory disorders: a nursing case report.
The Journal of international medical researchBAG3 mutation in a patient with atypical phenotypes of myofibrillar myopathy and Charcot-Marie-Tooth disease.
Genes & genomicsA Roma founder BIN1 mutation causes a novel phenotype of centronuclear myopathy with rigid spine.
NeurologySEPN1-related Rigid Spine Muscular Dystrophy.
Indian journal of pediatricsMuscular MRI-based algorithm to differentiate inherited myopathies presenting with spinal rigidity.
European radiologyRigid spine syndrome associated with sensory-motor axonal neuropathy resembling Charcot-Marie-Tooth disease is characteristic of Bcl-2-associated athanogene-3 gene mutations even without cardiac involvement.
Muscle & nerve[Diagnostic orientation of « Rigid spine » familial case with whole body muscle MRI].
Medecine sciences : M/STargeted next generation sequencing identifies two novel mutations in SEPN1 in rigid spine muscular dystrophy 1.
Oncotarget[Correlation between thigh muscle magnetic resonance imaging findings and clinical features of congenital muscular dystrophies: a preliminary study].
Zhonghua er ke za zhi = Chinese journal of pediatricsHypo- and Hyper-Assembly Diseases of RNA-Protein Complexes.
Trends in molecular medicineRigid Spine Syndrome among Children in Oman.
Sultan Qaboos University medical journalAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome da coluna rígida.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome da coluna rígida
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Novel missense variants in CFL2 affect F-actin depolymerisation and expand the disease spectrum of CFL2-related myopathy.
- New Clinical Phenotype in a Child Presenting With an FHL1 Mutation.
- HMGCS1 variants cause rigid spine syndrome amenable to mevalonic acid treatment in an animal model.
- SEPN1 Related Myopathy Presenting as Chronic Respiratory Insufficiency.
- The FHL1 myopathy spectrum revisited: a literature review and report of two new patients.Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology· 2024· PMID 40017287mais citado
- Genotype-phenotype correlation in recessive DNAJB4 myopathy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:97244(Orphanet)
- MONDO:0019951(MONDO)
- GARD:4723(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q29982088(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
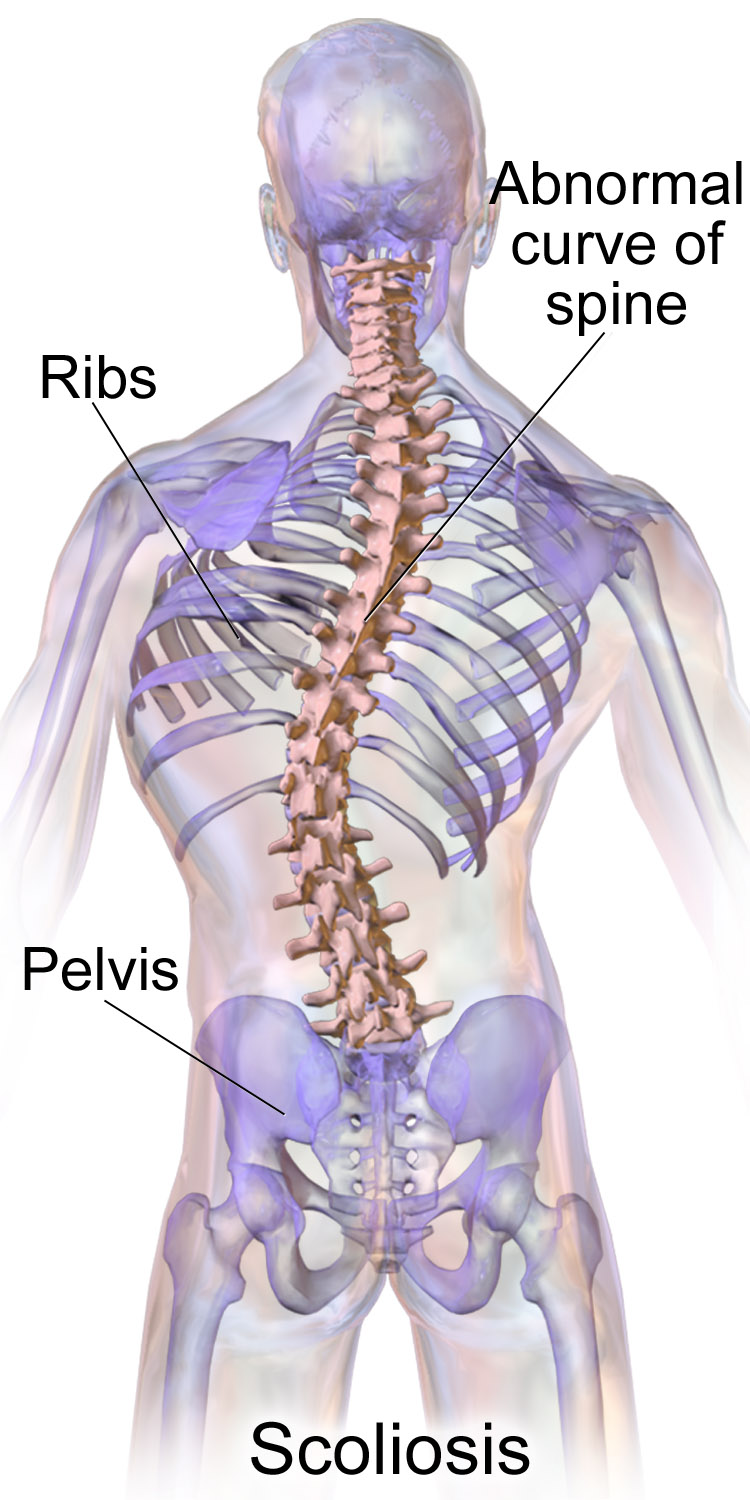
Síndrome da coluna rígida
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata