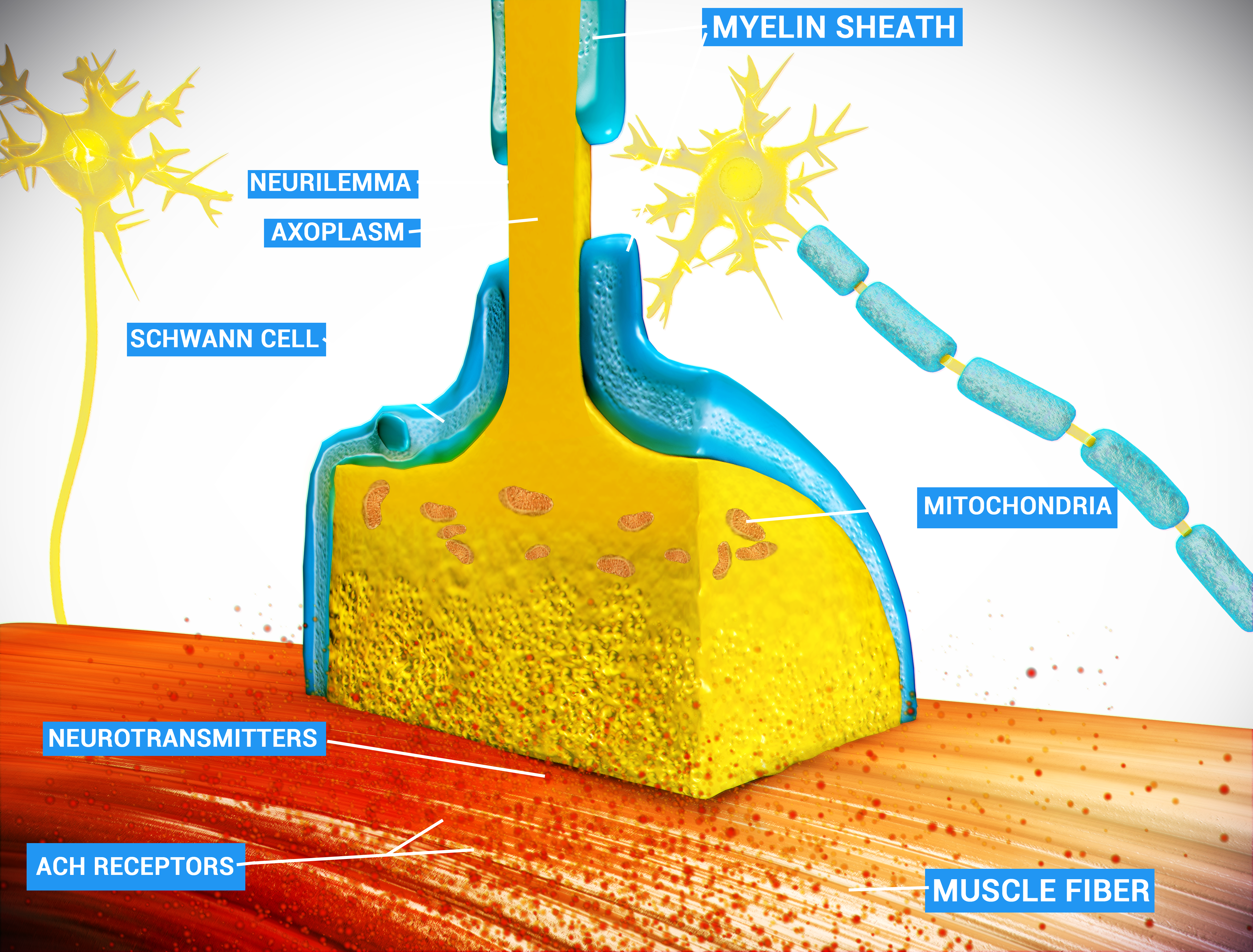
Condições caracterizadas por transmissão prejudicada de impulsos na junção neuromuscular. Isto pode resultar de distúrbios que afetam a função do receptor, a função da membrana pré ou pós-sináptica ou a atividade da acetilcolinesterase. A maioria das doenças nesta categoria está associada a doenças autoimunes, tóxicas ou hereditárias.
Introdução
O que você precisa saber de cara
Condições caracterizadas por transmissão prejudicada de impulsos na junção neuromuscular. Isto pode resultar de distúrbios que afetam a função do receptor, a função da membrana pré ou pós-sináptica ou a atividade da acetilcolinesterase. A maioria das doenças nesta categoria está associada a doenças autoimunes, tóxicas ou hereditárias.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 82 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 229 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
27 genes identificados com associação a esta condição.
Calcium sensor that plays a key role in processes such as endoplasmic reticulum (ER)-Golgi vesicular transport, endosomal biogenesis or membrane repair. Acts as an adapter that bridges unrelated proteins or stabilizes weak protein-protein complexes in response to calcium: calcium-binding triggers exposure of apolar surface, promoting interaction with different sets of proteins thanks to 3 different hydrophobic pockets, leading to translocation to membranes (PubMed:20691033, PubMed:25667979). Inv
Endoplasmic reticulum membraneCytoplasmic vesicle, COPII-coated vesicle membraneCytoplasmNucleusEndosome
Broad-specificity nucleoside phosphate kinase involved in cellular nucleotide homeostasis by catalyzing nucleoside-phosphate interconversions. Similar to other adenylate kinases, preferentially catalyzes the phosphorylation of the nucleoside monophosphate AMP with ATP as phosphate donor to produce ADP. In vitro, can also catalyze the phosphorylation of CMP, dAMP and dCMP and use GTP as an alternate phosphate donor. Moreover, exhibits a diphosphate kinase activity, producing ATP, CTP, GTP, UTP, T
CytoplasmNucleusCell projection, cilium, flagellum
Spermatogenic failure 89
An autosomal recessive male infertility disorder due to severely reduced progressive motility of sperm.
Binding to cells via a high affinity receptor, laminin is thought to mediate the attachment, migration and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components. As a subunit of laminin-1 (also known as laminin-111 or EHS laminin), it is involved in the stimulation of agrin-induced receptor clustering through a MuSK-independent pathway
Secreted, extracellular space, extracellular matrix, basement membrane
Pore-forming subunit of Nav1.4, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membrane
Paramyotonia congenita
An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP.
Postsynaptic protein required for clustering of nicotinic acetylcholine receptors (nAChRs) at the neuromuscular junction. It may link the receptor to the underlying postsynaptic cytoskeleton, possibly by direct association with actin or spectrin
Cell membranePostsynaptic cell membraneCytoplasm, cytoskeleton
Myasthenic syndrome, congenital, 11, associated with acetylcholine receptor deficiency
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS11 is an autosomal recessive disorder of postsynaptic neuromuscular transmission, due to deficiency of AChR at the endplate that results in low amplitude of the miniature endplate potential and current.
Mediates SOST-dependent inhibition of bone formation. Functions as a specific facilitator of SOST-mediated inhibition of Wnt signaling. Plays a key role in the formation and the maintenance of the neuromuscular junction (NMJ), the synapse between motor neuron and skeletal muscle. Directly binds AGRIN and recruits it to the MUSK signaling complex. Mediates the AGRIN-induced phosphorylation of MUSK, the kinase of the complex. The activation of MUSK in myotubes induces the formation of NMJ by regul
Cell membrane
Cenani-Lenz syndactyly syndrome
A congenital malformation syndrome defined as complete and complex syndactyly of the hands combined with malformations of the forearm bones and similar manifestations in the lower limbs. It is characterized by fusion and disorganization of metacarpal and phalangeal bones, radius and ulnar shortening, radioulnar synostosis, and severe syndactyly of hands and feet.
t-SNARE involved in the molecular regulation of neurotransmitter release. May play an important role in the synaptic function of specific neuronal systems. Associates with proteins involved in vesicle docking and membrane fusion. Regulates plasma membrane recycling through its interaction with CENPF. Modulates the gating characteristics of the delayed rectifier voltage-dependent potassium channel KCNB1 in pancreatic beta cells
Cytoplasm, perinuclear regionCell membraneSynapse, synaptosomePhotoreceptor inner segment
Developmental and epileptic encephalopathy 117
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE117 is an autosomal dominant form characterized by global developmental delay, delayed walking or inability to walk, intellectual disability, and early-onset seizures. Variable neurological symptoms are muscular hypotonia, movement disorders (ataxia, dystonia or tremor), cerebral visual impairment, and brain volume loss.
Part of the UDP-N-acetylglucosamine transferase complex that operates in the biosynthetic pathway of dolichol-linked oligosaccharides, the glycan precursors employed in protein asparagine (N)-glycosylation. The assembly of dolichol-linked oligosaccharides begins on the cytosolic side of the endoplasmic reticulum membrane and finishes in its lumen. The sequential addition of sugars to dolichol pyrophosphate produces dolichol-linked oligosaccharides containing fourteen sugars, including two GlcNAc
Endoplasmic reticulum membrane
Myasthenic syndrome, congenital, 15
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness.
Catalyzes the reversible synthesis of acetylcholine (ACh) from acetyl CoA and choline at cholinergic synapses
Myasthenic syndrome, congenital, 6, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive.
Serine peptidase whose precise substrate specificity remains unclear (PubMed:16143824, PubMed:16385448, PubMed:28726805). Does not cleave peptides after a arginine or lysine residue (PubMed:16143824). Regulates trans-Golgi network morphology and sorting by regulating the membrane binding of the AP-1 complex (PubMed:23321636). May play a role in the regulation of synaptic vesicle exocytosis (PubMed:24610330)
Cytoplasm, cytosolGolgi apparatus, trans-Golgi networkCytoplasm, cytoskeletonGolgi apparatusNucleus
Hypotonia-cystinuria syndrome
Characterized generalized hypotonia at birth, nephrolithiasis, growth hormone deficiency, minor facial dysmorphism, failure to thrive, followed by hyperphagia and rapid weight gain in late childhood.
High-affinity Na(+)-coupled choline transmembrane symporter (PubMed:11027560, PubMed:11068039, PubMed:12237312, PubMed:12969261, PubMed:17005849, PubMed:23132865, PubMed:23141292, PubMed:27569547). Functions as an electrogenic, voltage-dependent transporter with variable charge/choline stoichiometry (PubMed:17005849). Choline uptake and choline-induced current is also Cl(-)-dependent where Cl(-) is likely a regulatory ion rather than cotransported ion (PubMed:11068039, PubMed:12237312, PubMed:17
Presynaptic cell membraneCell projection, axonEarly endosome membraneCytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Neuronopathy, distal hereditary motor, autosomal dominant 7
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMND7 is characterized by onset in the second decade of progressive distal muscle wasting and weakness affecting the upper and lower limbs and resulting in walking difficulties and hand grip. There is significant muscle atrophy of the hands and lower limbs. The disorder is associated with vocal cord paresis due to involvement of the tenth cranial nerve.
UDP-N-acetylglucosamine--dolichyl-phosphate N-acetylglucosaminephosphotransferase that operates in the biosynthetic pathway of dolichol-linked oligosaccharides, the glycan precursors employed in protein asparagine (N)-glycosylation. The assembly of dolichol-linked oligosaccharides begins on the cytosolic side of the endoplasmic reticulum membrane and finishes in its lumen. The sequential addition of sugars to dolichol pyrophosphate produces dolichol-linked oligosaccharides containing fourteen su
Endoplasmic reticulum membrane
Congenital disorder of glycosylation 1J
A form of congenital disorder of glycosylation, a multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions.
Rate-limiting enzyme of the hexosamine biosynthetic pathway (HBP) that catalyzes the formation of glucosamine-6-phosphate from fructose-6-phosphate and glutamine, thereby controlling the flux of glucose into this pathway (PubMed:32019926, PubMed:35229715). Inhibited by UDP-N-acetylglucosamine (UDP-GlcNAc) through a feedback loop (PubMed:32019926, PubMed:35229715). Fine-tunes the metabolic fluctuations of UDP-GlcNAc and its impacts on hyaluronan synthesis during tissue remodeling (PubMed:26887390
Myasthenic syndrome, congenital, 12
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS12 is characterized by onset of proximal muscle weakness in the first decade. Individuals with this condition have a recognizable pattern of weakness of shoulder and pelvic girdle muscles, and sparing of ocular or facial muscles. EMG classically shows a decremental response to repeated nerve stimulation, a sign of neuromuscular junction dysfunction. Affected individuals show a favorable response to acetylcholinesterase (AChE) inhibitors.
Anchors the catalytic subunits of asymmetric AChE to the synaptic basal membrane, and is therefore involved in the down-regulation of colinergic synaptic transmission
Synapse
Myasthenic syndrome, congenital, 5
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS5 inheritance is autosomal recessive.
Involved in cell-matrix and cell-cell adhesion interactions that are required for normal development. May participate in the linkage between muscle fiber and basement membrane. May play a role in endochondral ossification of bone and branching morphogenesis of lung. Binds heparin. At neuromuscular junctions, may play a role in acetylcholine receptor clustering (PubMed:26626625)
Cell membranePostsynaptic cell membrane
Myasthenic syndrome, congenital, 19
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort.
After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane
Postsynaptic cell membraneCell membrane
Myosins are actin-based motor molecules with ATPase activity. Unconventional myosins serve in intracellular movements. Regulates Rho by stimulating its GTPase activity in neurons. Required for the regulation of neurite branching and motor neuron axon guidance (By similarity)
MembraneCytoplasmSynapseCell projection, growth cone
Myasthenic syndrome, congenital, 24, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features include easy fatigability and muscle weakness. CMS24 inheritance is autosomal recessive.
Electrogenic antiporter that exchanges one cholinergic neurotransmitter, acetylcholine or choline, with two intravesicular protons across the membrane of synaptic vesicles. Uses the electrochemical proton gradient established by the V-type proton-pump ATPase to store neurotransmitters inside the vesicles prior to their release via exocytosis (By similarity) (PubMed:20225888, PubMed:8910293). Determines cholinergic vesicular quantal size at presynaptic nerve terminals in developing neuro-muscular
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Myasthenic syndrome, congenital, 21, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS21 is an autosomal recessive, pre-synaptic form characterized by ptosis, ophthalmoplegia, fatigable weakness, apneic crises, and deterioration of symptoms in cold water. Learning difficulties and left ventricular dysfunction may be present in some patients.
Probable muscle-intrinsic activator of MUSK that plays an essential role in neuromuscular synaptogenesis. Acts in aneural activation of MUSK and subsequent acetylcholine receptor (AchR) clustering in myotubes. Induces autophosphorylation of MUSK
Cell membraneSynapse
Myasthenic syndrome, congenital, 10
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS10 is an autosomal recessive, post-synaptic form characterized by a typical 'limb girdle' pattern of muscle weakness with small, simplified neuromuscular junctions but normal acetylcholine receptor and acetylcholinesterase function.
After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane
Postsynaptic cell membraneCell membrane
Multiple pterygium syndrome, lethal type
Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent.
Involved in the targeting and/or fusion of transport vesicles to their target membrane
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membraneSynapse, synaptosomeCytoplasmic vesicle membraneMitochondrion outer membrane
Spastic ataxia 1, autosomal dominant
An autosomal dominant form of spastic ataxia, a progressive neurodegenerative disorder characterized by lower-limb spasticity and generalized ataxia with dysarthria, impaired ocular movements, and gait disturbance.
Receptor tyrosine kinase which plays a central role in the formation and the maintenance of the neuromuscular junction (NMJ), the synapse between the motor neuron and the skeletal muscle (PubMed:25537362). Recruitment of AGRIN by LRP4 to the MUSK signaling complex induces phosphorylation and activation of MUSK, the kinase of the complex. The activation of MUSK in myotubes regulates the formation of NMJs through the regulation of different processes including the specific expression of genes in s
Postsynaptic cell membrane
Myasthenic syndrome, congenital, 9, associated with acetylcholine receptor deficiency
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS9 is a disorder of postsynaptic neuromuscular transmission, due to deficiency of AChR at the endplate that results in low amplitude of the miniature endplate potential and current.
Depending on alternative splicing and post-translational modifications, it has a role in different processes, including neuromuscular junction formation and maintenance, and regulation of neurite outgrowth (By similarity). Also involved in positive regulation of cartilage formation through alpha-dystroglycan binding and up-regulation of SOX9 (PubMed:26290588) Heparan sulfate basal lamina glycoprotein that plays a central role in the formation and the maintenance of the neuromuscular junction (NM
Secreted, extracellular space, extracellular matrixSynapseCell membrane
Myasthenic syndrome, congenital, 8
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS8 is an autosomal recessive disease characterized by prominent defects of both the pre- and postsynaptic regions. Affected individuals have onset of muscle weakness in early childhood; the severity of the weakness and muscles affected is variable.
After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane
Postsynaptic cell membraneCell membrane
Myasthenic syndrome, congenital, 2A, slow-channel
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS2A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane.
Exhibits calcium-dependent phospholipid and inositol polyphosphate binding properties (By similarity). May have a regulatory role in the membrane interactions during trafficking of synaptic vesicles at the active zone of the synapse (By similarity). Plays a role in dendrite formation by melanocytes (PubMed:23999003)
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membraneCytoplasmic vesicle, secretory vesicle, chromaffin granule membraneCytoplasm
Myasthenic syndrome, congenital, 7A, presynaptic, and distal motor neuropathy, autosomal dominant
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS7A is an autosomal dominant, presynaptic disorder resembling Lambert-Eaton myasthenic syndrome. Affected individuals have a variable degree of proximal and distal limb weakness, muscle fatigue that improves with rest, mild gait difficulties, and reduced or absent deep tendon reflexes.
Mitochondrial electroneutral antiporter that exports citrate from the mitochondria into the cytosol in exchange for malate (PubMed:26870663, PubMed:29031613, PubMed:29238895, PubMed:39881208, PubMed:38937634). Also able to mediate the exchange of citrate for isocitrate, phosphoenolpyruvate, cis-aconitate and to a lesser extent trans-aconitate, maleate and succinate (PubMed:29031613). Substrate exchange across the membrane occurs consecutively with one substrate being transported first, then diss
Mitochondrion inner membrane
Combined D-2- and L-2-hydroxyglutaric aciduria
An autosomal recessive neurometabolic disorder characterized by neonatal-onset encephalopathy with severe muscular weakness, intractable seizures, respiratory distress, and lack of psychomotor development resulting in early death. Brain imaging shows abnormalities including enlarged ventricles, delayed myelination, and germinal layer cysts.
Upon acetylcholine binding, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane Non functional acetylcholine receptor alpha subunit which is not integrated into functional acetylcholine-gated cation-selective channels
Postsynaptic cell membraneCell membrane
Multiple pterygium syndrome, lethal type
Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent.
Variantes genéticas (ClinVar)
242 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
72 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença da junção neuromuscular
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Mendelian randomization followed by single-cell RNA sequencing exploration identifies effector memory CD4+ T cells as a risk factor for myasthenia gravis.
Este estudo identificou um tipo específico de célula imunológica, as células T CD4+ de memória efetora, como um importante fator de risco para a Miastenia Gravis de início tardio (LOMG). Essas células foram encontradas em maior proporção em pacientes com LOMG e se expandem significativamente durante as crises da doença. Essa descoberta aprofunda a compreensão da patogênese da LOMG e pode abrir caminho para o desenvolvimento de novas estratégias de tratamento mais direcionadas.
🇧🇷 traduzidoRehabilitation in Neuromuscular Diseases: Best Turkish Practice Recommendations by Multidisciplinary Experts.
Este estudo turco preencheu uma lacuna importante ao desenvolver recomendações de reabilitação abrangentes para doenças neuromusculares (DNMs), incluindo as da junção neuromuscular (JNM), para pacientes de todas as idades. Uma equipe multidisciplinar de especialistas utilizou o método Delphi para criar 110 diretrizes detalhadas, que cobrem desde a gestão geral até prescrições específicas de exercícios. Essas recomendações são um guia prático essencial para médicos, otimizando o cuidado e a reabilitação de pacientes com condições como as da junção neuromuscular.
🇧🇷 traduzidoEarly prediction of refractory myasthenia gravis based on response to treatment within the first year of diagnosis.
Este estudo demonstra que é possível prever precocemente a Miastenia Gravis (MG) refratária, uma forma mais difícil de tratar, com base na resposta ao tratamento durante o primeiro ano de diagnóstico. Pacientes que não conseguem reduzir a dose de prednisona para menos de 20mg/dia e que necessitam iniciar outros imunossupressores orais nesse período têm um risco significativamente maior de desenvolver a doença refratária. Essa identificação precoce é crucial para médicos e pacientes, pois permite considerar intervenções mais avançadas ou novas terapias de forma mais ágil, visando melhores resultados.
🇧🇷 traduzidoCOVID-19-associated delayed-onset MuSK-positive myasthenia gravis presenting solely with respiratory failure: a case report.
Este artigo relata um caso raro de miastenia gravis MuSK-positiva, que se manifestou exclusivamente com insuficiência respiratória grave de início tardio, desencadeada por COVID-19. Para médicos, é crucial considerar esta doença neuromuscular em casos inexplicados de falha no desmame do ventilador — mesmo sem fraqueza muscular típica ou testes iniciais negativos. O diagnóstico precoce com anticorpos MuSK e o tratamento imunomodulador rápido (como troca de plasma) são vitais para uma recuperação rápida e podem salvar vidas.
🇧🇷 traduzidoSYT2-Related Disease: A Case-Based Review.
A doença ultrarrara relacionada ao gene SYT2 manifesta-se por fraqueza e atrofia muscular distal (principalmente nas pernas), deformidades nos pés e, em alguns casos, hipotonia neonatal (bebê "mole"). Este artigo revisa os aspectos clínicos e eletrofisiológicos, descrevendo um novo caso e comparando-o com outros 27, o que é crucial para médicos no diagnóstico e para pacientes na compreensão desta doença da junção neuromuscular, apesar de ser por vezes considerada uma neuropatia motora hereditária.
🇧🇷 traduzidoPublicações recentes
Mendelian randomization followed by single-cell RNA sequencing exploration identifies effector memory CD4+ T cells as a risk factor for myasthenia gravis.
COVID-19-associated delayed-onset MuSK-positive myasthenia gravis presenting solely with respiratory failure: a case report.
SYT2-Related Disease: A Case-Based Review.
Rehabilitation in Neuromuscular Diseases: Best Turkish Practice Recommendations by Multidisciplinary Experts.
Algorithm for jitter measurement in neuromuscular junction disease.
📚 EuropePMC5 artigos no totalmostrando 25
Mendelian randomization followed by single-cell RNA sequencing exploration identifies effector memory CD4+ T cells as a risk factor for myasthenia gravis.
MedicineCOVID-19-associated delayed-onset MuSK-positive myasthenia gravis presenting solely with respiratory failure: a case report.
Frontiers in immunologySYT2-Related Disease: A Case-Based Review.
Journal of clinical neuromuscular diseaseRehabilitation in Neuromuscular Diseases: Best Turkish Practice Recommendations by Multidisciplinary Experts.
Acta neurologica BelgicaAlgorithm for jitter measurement in neuromuscular junction disease.
Journal of electromyography and kinesiology : official journal of the International Society of Electrophysiological KinesiologyEarly prediction of refractory myasthenia gravis based on response to treatment within the first year of diagnosis.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyBilateral horizontal gaze palsy, esotropia, and diplopia secondary to anti-Hu paraneoplastic syndrome.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusThe Effect of Apomorphine Therapy in the Coexistence of Parkinson's Disease and Myasthenia Gravis: A Case Report and Review of the Literature.
Journal of clinical practice and researchMachine learning strategy for identifying altered gut microbiomes for diagnostic screening in myasthenia gravis.
Frontiers in microbiologyCOVID-19 vaccination-related autoimmune hepatitis-a perspective.
Frontiers in pharmacologySeronegative Ocular Myasthenia Gravis in an Older Woman With Transient Dizziness and Diplopia.
CureusA Common CHAT Gene Mutation of Congenital Myasthenic Syndrome Found in Kadazandusun Children.
Journal of pediatric geneticsAcquired myasthenia gravis with concurrent polymyositis and myocarditis secondary to a thymoma in a dog.
Open veterinary journalMyasthenia Gravis-An Analysis of Multimodal Evoked Potentials.
Brain sciencesAge at sampling and sex distribution of AChRAb vs. MuSKAb myasthenia gravis in a large Greek population.
Clinical neurology and neurosurgeryDual attack: targeting the rare co-occurrence of myasthenia gravis and Graves' disease with radioactive iodine therapy.
Endocrinology, diabetes & metabolism case reportsSocial, professional and neuropsychiatric outcomes in patients with myasthenia gravis.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyParaneoplastic Syndromes in Neuro-ophthalmology.
Continuum (Minneapolis, Minn.)Medication adherence in patients with myasthenia gravis in Brazil: a cross-sectional study.
Acta neurologica BelgicaClinical neurophysiology of neuromuscular junction disease.
Handbook of clinical neurologySingle fiber electromyography.
Handbook of clinical neurologyClinical Image: Leriche Syndrome.
Journal of acute medicineAn investigation on the effects of carbamazepine and sodium valproate on neuromuscular transmission.
Acta neurologica BelgicaAnalysis of TNF-related apoptosis-inducing ligand and receptors and implications in thymus biology and myasthenia gravis.
Neuromuscular disorders : NMDCost-minimization analysis comparing intravenous immunoglobulin with plasma exchange in the management of patients with myasthenia gravis.
Muscle & nerveAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença da junção neuromuscular.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença da junção neuromuscular
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Mendelian randomization followed by single-cell RNA sequencing exploration identifies effector memory CD4+ T cells as a risk factor for myasthenia gravis.
- Rehabilitation in Neuromuscular Diseases: Best Turkish Practice Recommendations by Multidisciplinary Experts.
- Early prediction of refractory myasthenia gravis based on response to treatment within the first year of diagnosis.Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology· 2025· PMID 40281192mais citado
- COVID-19-associated delayed-onset MuSK-positive myasthenia gravis presenting solely with respiratory failure: a case report.
- SYT2-Related Disease: A Case-Based Review.
- Algorithm for jitter measurement in neuromuscular junction disease.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:98491(Orphanet)
- MONDO:0020124(MONDO)
- GARD:19473(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7002430(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar