A acidúria 2-hidroxiglutárica é um grupo de distúrbios neurometabólicos com amplo espectro clínico que varia de apresentações neonatais graves a formas progressivas e casos assintomáticos, caracterizados bioquimicamente por níveis aumentados de ácido 2-hidroxiglutárico no plasma, líquido cefalorraquidiano e urina.
Introdução
O que você precisa saber de cara
A acidúria 2-hidroxiglutárica é um grupo de distúrbios neurometabólicos com amplo espectro clínico que varia de apresentações neonatais graves a formas progressivas e casos assintomáticos, caracterizados bioquimicamente por níveis aumentados de ácido 2-hidroxiglutárico no plasma, líquido cefalorraquidiano e urina.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 24 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 70 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Triagem neonatal (Teste do Pezinho)
A triagem neonatal permite diagnóstico precoce e início imediato do tratamento.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Plays a role in intermediary metabolism and energy production (PubMed:19228619, PubMed:22416140). It may tightly associate or interact with the pyruvate dehydrogenase complex (PubMed:19228619, PubMed:22416140)
Mitochondrion
D-2-hydroxyglutaric aciduria 2
A neurometabolic disorder causing developmental delay, epilepsy, hypotonia, and dysmorphic features. Both a mild and a severe phenotype exist. The severe phenotype is homogeneous and is characterized by early infantile-onset epileptic encephalopathy and cardiomyopathy. The mild phenotype has a more variable clinical presentation. Diagnosis is based on the presence of an excess of D-2-hydroxyglutaric acid in the urine.
Catalyzes the oxidation of D-2-hydroxyglutarate (D-2-HG) to alpha-ketoglutarate (PubMed:15070399, PubMed:15609246, PubMed:16037974, PubMed:20020533, PubMed:33431826). Also catalyzes the oxidation of other D-2-hydroxyacids, such as D-malate (D-MAL) and D-lactate (D-LAC) (PubMed:33431826). Exhibits high activities towards D-2-HG and D-MAL but a very weak activity towards D-LAC (PubMed:33431826)
Mitochondrion
D-2-hydroxyglutaric aciduria 1
A rare recessive neurometabolic disorder causing developmental delay, epilepsy, hypotonia, and dysmorphic features. Both a mild and a severe phenotype exist. The severe phenotype is homogeneous and is characterized by early infantile-onset epileptic encephalopathy and cardiomyopathy. The mild phenotype has a more variable clinical presentation. Diagnosis is based on the presence of an excess of D-2-hydroxyglutaric acid in the urine.
Mitochondrial electroneutral antiporter that exports citrate from the mitochondria into the cytosol in exchange for malate (PubMed:26870663, PubMed:29031613, PubMed:29238895, PubMed:39881208, PubMed:38937634). Also able to mediate the exchange of citrate for isocitrate, phosphoenolpyruvate, cis-aconitate and to a lesser extent trans-aconitate, maleate and succinate (PubMed:29031613). Substrate exchange across the membrane occurs consecutively with one substrate being transported first, then diss
Mitochondrion inner membrane
Combined D-2- and L-2-hydroxyglutaric aciduria
An autosomal recessive neurometabolic disorder characterized by neonatal-onset encephalopathy with severe muscular weakness, intractable seizures, respiratory distress, and lack of psychomotor development resulting in early death. Brain imaging shows abnormalities including enlarged ventricles, delayed myelination, and germinal layer cysts.
Mitochondrion
L-2-hydroxyglutaric aciduria
A rare autosomal recessive disorder clinically characterized by mild psychomotor delay in the first years of life, followed by progressive cerebellar ataxia, dysarthria and moderate to severe intellectual disability. Diagnosis is based on the presence of an excess of L-2-hydroxyglutaric acid in urine, blood and cerebrospinal fluid.
Variantes genéticas (ClinVar)
652 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 8 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
6 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Acidúria 2-hidroxiglutárica
Centros de Referência SUS
21 centros habilitados pelo SUS para Acidúria 2-hidroxiglutárica
Centros para Acidúria 2-hidroxiglutárica
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
NUPAD / Faculdade de Medicina UFMG
Av. Prof. Alfredo Balena, 189 - 5 andar - Centro, Belo Horizonte - MG, 30130-100 · CNES 2183226
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital de Clínicas da Universidade Federal de Pernambuco
Av. Prof. Moraes Rego, 1235 - Cidade Universitária, Recife - PE, 50670-901 · CNES 2561492
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital Universitário Onofre Lopes (HUOL)
Av. Nilo Peçanha, 620 - Petrópolis, Natal - RN, 59012-300 · CNES 2408570
Atenção Especializada
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Instituto da Criança e do Adolescente (ICr-HCFMUSP)
Av. Dr. Enéas Carvalho de Aguiar, 647 - Cerqueira César, São Paulo - SP, 05403-000 · CNES 2081695
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
[A case of L-2-hydroxyglutaric aciduria diagnosed with involuntary movements, in which improvement in motor symptoms was achieved following treatment].
A 49-year-old female presented with the primary complaint of hand tremors. Neurological examination on admission revealed signs of cognitive impairment, bulbar palsy, dystonia, cerebellar ataxia, and pyramidal tract disease. T2-weighted brain MRI revealed hyperintense signals in the subcortical white matter, basal ganglia, and cerebellar dentate nucleus, with no atrophy of the brainstem or corpus callosum. Urinary organic acid analysis revealed elevated 2-hydroxyglutaric acid levels. Although the optical isomers could not be distinguished, L-2-hydroxyglutaric aciduria was diagnosed based on the disease course, symptoms, and characteristic MRI findings. The patient was started on riboflavin-enriched compounds and levocarnitine, resulting in an improvement in the Scale for the Assessment and Rating of Ataxia (SARA) score from 21 to 15 after six months. The case suggests that symptoms in adult patients who have not been treated for a long time can be improved by appropriate diagnosis based on neurological presentation, characteristic MRI findings, and intervention.
Successful Treatment of 2-hydroxyglutaric Aciduria Diagnosed in Adulthood, Three Decades After the Onset: A Case Report and Literature Review.
2-Hydroxyglutaric aciduria is a rare genetic metabolic disorder, especially in Japan. Although magnetic resonance images show characteristic abnormalities in the subcortical white matter, some cases have been diagnosed in adulthood, namely many decades after onset. We herein report the case of a bedridden 37-year-old 2-hydroxyglutaric aciduria male patient who was diagnosed three decades after onset. Despite this, combination treatment with riboflavin and levocarnitine improved his condition, thus allowing him to walk by himself. Considering our case and the previous literature, we emphasize the importance of correctly diagnosing and treating patients with 2-hydroxyglutaric aciduria.
A Pathogenic L2HGDH Variant Impairs Mitochondrial Targeting and Enzyme Function in L-2-Hydroxyglutaric Aciduria: Clinical and Functional Evidence from Two Affected Siblings.
L-2-hydroxyglutaric aciduria (L2HGA) is a rare autosomal recessive neurometabolic disorder caused by biallelic loss-of-function variants in the L-2-hydroxyglutarate dehydrogenase (L2HGDH) gene, leading to accumulation of L-2-hydroxyglutarate in the brain and other tissues. While various variants have been reported, the pathogenic mechanism of specific variants remains unclear. In this study, we aimed to investigate the molecular consequences of the c.905C>T p.(Pro302Leu) variant, identified in two siblings with typical symptoms of L2HGA, by analyzing its effects on protein localization and enzymatic activity in a cell model. HA-tagged wild-type and p.(Pro302Leu) mutant L2HGDH constructs were overexpressed in HEK293T cells. We assessed protein expression, subcellular localization, and enzymatic activity using Western blot analysis, immunofluorescence microscopy, and a specific enzyme assay measuring 2,6-dichloroindophenol (DCIP) reduction to assess L2HGDH enzymatic activity. Western blotting showed that wild-type L2HGDH exists primarily in the processed, mature mitochondrial form, whereas the p.(Pro302Leu) mutant remained largely in the unprocessed precursor form. Immunofluorescence and differential centrifugation revealed that wild-type protein localized to mitochondria, while the mutant protein accumulated in the cytoplasm in a diffuse or punctate pattern. Enzyme activity assays demonstrated that the mutant retained <30% of wild-type activity. These findings indicate that the p.(Pro302Leu) variant leads to aggregation of mislocalized protein, thereby impairing L2HGDH function rather than decreasing enzymatic function. This study provides clinical and molecular evidence supporting the pathogenicity of this previously reported mutation and highlights the importance of mitochondrial import for enzyme functionality in L2HGA.
Genetic, neuroimaging, and clinical characteristics of a cohort of individuals with L-2-hydroxyglutaric aciduria from Türkiye.
L-2-hydroxyglutaric aciduria (L2HGA) is a hereditary metabolic disorder characterized by the accumulation of L-2-hydroxyglutaric acid in body fluids, particularly in cerebrospinal fluid, which disrupts neuron function in the central nervous system and triggers oxidative stress. It can cause seizures, developmental disorders, and behavioral abnormalities. The study retrospectively evaluated the demographic information, initial symptoms, clinical characteristics, cranial magnetic resonance imaging (MRI) findings, and post-treatment biochemical changes of 10 cases diagnosed with L2HGA. The study included five paediatric and five adult cases with a molecular diagnosis of L2HGA. The mean age at diagnosis was 10.1 years. Convulsion was identified as the primary presenting symptom in 70 % of cases. We identified intellectual disability in 80 % of our cases. In addition to the classic cranial MRI findings of subcortical white matter involvement, basal ganglia involvement was detected in 60 % of cases. We found that 2-hydroxyglutaric acid levels in urine organic acid analysis were significantly decreased riboflavin and carnitine post-treatment, with a mean decrease of 133.89 ± 101.43 mmol/mol creatinine (p=0.017). The most common missense variant identified in the L2HGDH gene was c.905C>T (p.Pro302Leu), occurring at a frequency of 50 % (5/10). The cases did not report significant improvement in their symptoms with treatment. L2HGA is a rare metabolic disorder that is more common in communities where consanguineous marriages are prevalent. Early diagnosis enables early treatment and protection of the brain from oxidative stress. As more cases are reported publicly, studies on genotype-phenotype relationships will yield more significant findings.
Riboflavin treatment in L-2-hydroxyglutaric aciduria: report on a pediatric patient and literature review.
L-2-hydroxyglutaric aciduria (L-2-HGA, #236,792) is an autosomal recessive neurodegenerative disorder caused by the deficiency of L-2-hydroxyglutarate dehydrogenase, a flavin adenine dinucleotide (FAD)-dependent enzyme, due to biallelic pathogenic variants in the L2HGDH gene. The present study described the patient with L2HGA presenting with a slight psychomotor delay, epilepsy from 5 years of age, non-progressive cerebellar ataxia, and mild to moderate intellectual disability during 10 years of follow-up. Two different heterozygous variants in the L2HGDH gene were identified in the patient: a known substitution c.829C > T(p.Arg277*) and a novel substitution c.1196 + 1G > A corresponding with significantly increased urinary L-2-hydroxyglutarate (L2HG) excretion. A 6-month period of treatment with riboflavin (100 mg/day) was implemented with no clinical nor biochemical effect.
Publicações recentes
Subtle clinical and radiological findings as potential indicators of CNS tumors in L-2-hydroxyglutaric aciduria: a literature review.
Successful Treatment of 2-hydroxyglutaric Aciduria Diagnosed in Adulthood, Three Decades After the Onset: A Case Report and Literature Review.
🥉 Relato de casoA Pathogenic L2HGDH Variant Impairs Mitochondrial Targeting and Enzyme Function in L-2-Hydroxyglutaric Aciduria: Clinical and Functional Evidence from Two Affected Siblings.
Genetic, neuroimaging, and clinical characteristics of a cohort of individuals with L-2-hydroxyglutaric aciduria from Türkiye.
🥉 Relato de casoL-2-Hydroxyglutaric Aciduria Complicated by Cerebral Neoplasm.
📚 EuropePMC204 artigos no totalmostrando 53
Successful Treatment of 2-hydroxyglutaric Aciduria Diagnosed in Adulthood, Three Decades After the Onset: A Case Report and Literature Review.
Internal medicine (Tokyo, Japan)A Pathogenic L2HGDH Variant Impairs Mitochondrial Targeting and Enzyme Function in L-2-Hydroxyglutaric Aciduria: Clinical and Functional Evidence from Two Affected Siblings.
GenesGenetic, neuroimaging, and clinical characteristics of a cohort of individuals with L-2-hydroxyglutaric aciduria from Türkiye.
Journal of pediatric endocrinology & metabolism : JPEMRiboflavin treatment in L-2-hydroxyglutaric aciduria: report on a pediatric patient and literature review.
Journal of applied genetics[A case of L-2-hydroxyglutaric aciduria diagnosed with involuntary movements, in which improvement in motor symptoms was achieved following treatment].
Rinsho shinkeigaku = Clinical neurologyCutaneous manifestations in D-2-hydroxyglutaric aciduria type 2 and response to enasidenib therapy.
JAAD case reportsProtective effects of the PPAR agonist bezafibrate against disruption of redox and energy homeostasis, neuronal death, astroglial reactivity, and neuroinflammation induced in vivo by D-2-hydroxyglutaric acid in rat brain.
European journal of pharmacologyFocal dystonia in an adult with L-2- hydroxyglutaric aciduria.
Saudi medical journalL-2-hydroxyglutaric aciduria: a report of clinical, radiological, and genetic characteristics of two siblings from Egypt.
NeurocaseMalignant glioma in L-2-Hydroxyglutaric Aciduria: thorough molecular characterization of a case and literature review.
Free neuropathologyLate-onset cerebellar ataxia and a new frameshift L2HGDH mutation in a Chinese adult with L-2-hydroxyglutaric aciduria: a case report.
Acta neurologica BelgicaMoyamoya syndrome in a patient with D-2-hydroxyglutaric aciduria type II: a rare association.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryL-2 hydroxyglutaric aciduria: report of a Mexican-Mayan patient with the mutation c.569C>T and response to vitamin supplements and levocarnitine.
Tremor and other hyperkinetic movements (New York, N.Y.)Structure and biochemical characterization of l-2-hydroxyglutarate dehydrogenase and its role in the pathogenesis of l-2-hydroxyglutaric aciduria.
The Journal of biological chemistryLoss of function variants in L2HGDH gene causing L-2-hydroxyglutaric aciduria.
Acta neurologica BelgicaSeparation and quantification of the urinary enantiomers of 2-hydroxyglutaric acid by capillary electrophoresis with capacitively coupled contactless conductivity detection: Application to the diagnosis of D- and L-2-hydroxyglutaric aciduria.
Journal of separation scienceL-2-hydroxyglutaric aciduria - review of literature and case series.
Annals of medicine and surgery (2012)An intermediate phenotype in IDH related enchondromatosis spectrum.
European journal of medical geneticsL-2-Hydroxyglutaric Acid Administration to Neonatal Rats Elicits Marked Neurochemical Alterations and Long-Term Neurobehavioral Disabilities Mediated by Oxidative Stress.
Neurotoxicity researchDisruption of mitochondrial bioenergetics, calcium retention capacity and cell viability caused by D-2-hydroxyglutaric acid in the heart.
BiochimieAcute myeloid leukemia and dilated cardiomyopathy in a pediatric patient with D-2-hydroxyglutaric aciduria type I.
American journal of medical genetics. Part AA novel protein truncating mutation in L2HGDH causes L-2-hydroxyglutaric aciduria in a consanguineous Pakistani family.
Metabolic brain diseasePostural tremor in L-2-hydroxyglutaric aciduria is associated with cerebellar atrophy.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyBilateral subdural hematomas and retinal hemorrhages mimicking nonaccidental trauma in a patient with D-2-hydroxyglutaric aciduria.
JIMD reportsThe Wide Phenotypic Spectrum of L-2 Hydroxyglutaric Aciduria in Adults.
Movement disorders clinical practiceEvaluation of clinical, neuroradiologic, and genotypic features of patients with L-2-hydroxyglutaric aciduria.
Turk pediatri arsiviA case report of an intermediate phenotype between congenital myasthenic syndrome and D-2- and L-2-hydroxyglutaric aciduria due to novel SLC25A1 variants.
BMC neurologyA Case Report of Chronic Progressive Pancerebellar Syndrome with Leukoencephalopathy:L-2 Hydroxyglutaric Aciduria.
Movement disorders clinical practiceClassic Imaging Features of L-2-Hydroxyglutaric Aciduria in Young Adult Presenting as Seizures Associated with Fever.
Annals of Indian Academy of NeurologyA 7-year-old Boy with Hand Tremors and a Novel Mutation for L-2-hydroxyglutaric Aciduria.
Balkan journal of medical genetics : BJMGD-2-Hydroxyglutaric Aciduria with Enchondromatosis and Angiokeratoma Circumscriptum.
CureusCerebral neoplasm in L-2-hydroxyglutaric aciduria: two different presentations.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryInborn errors of metabolite repair.
Journal of inherited metabolic diseaseBiochemical characterization of human D-2-hydroxyglutarate dehydrogenase and two disease related variants reveals the molecular cause of D-2-hydroxyglutaric aciduria.
Biochimica et biophysica acta. Proteins and proteomics2-Hydroxyglutaric aciduria as a cause for seizure-like episodes in a domestic shorthair cat.
JFMS open reportsD-2-hydroxyglutaric aciduria Type I: Functional analysis of D2HGDH missense variants.
Human mutationTwo novel L2HGDH mutations identified in a rare Chinese family with L-2-hydroxyglutaric aciduria.
BMC medical geneticsComputational approach to unravel the impact of missense mutations of proteins (D2HGDH and IDH2) causing D-2-hydroxyglutaric aciduria 2.
Metabolic brain diseaseA novel compound heterozygous mutation of the L2HGDH gene in a Chinese boy with L-2-hydroxyglutaric aciduria: case report and literature review.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyWidespread and debilitating hemangiomas in a patient with enchondromatosis and D-2-hydroxyglutaric aciduria.
Skeletal radiologyMuscle Weakness, Cardiomyopathy, and L-2-Hydroxyglutaric Aciduria Associated with a Novel Recessive SLC25A4 Mutation.
JIMD reportsIdentification of novel L2HGDH mutation in a large consanguineous Pakistani family- a case report.
BMC medical geneticsExperimental Evidence that In Vivo Intracerebral Administration of L-2-Hydroxyglutaric Acid to Neonatal Rats Provokes Disruption of Redox Status and Histopathological Abnormalities in the Brain.
Neurotoxicity researchA novel homozygous SLC25A1 mutation with impaired mitochondrial complex V: Possible phenotypic expansion.
American journal of medical genetics. Part A[L-2-hydroxyglutaric aciduria caused by a new mutation in the L2HGDH gene].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaExperimental evidence of oxidative stress in patients with l-2-hydroxyglutaric aciduria and that l-carnitine attenuates in vitro DNA damage caused by d-2-hydroxyglutaric and l-2-hydroxyglutaric acids.
Toxicology in vitro : an international journal published in association with BIBRAClinical features and disease progression of L-2-hydroxyglutaric aciduria in 27 Staffordshire bull terriers.
The Veterinary recordMRI features in 17 patients with l2 hydroxyglutaric aciduria.
European journal of radiology openIn-vivo brain H1-MR-Spectroscopy identification and quantification of 2-hydroxyglutarate in L-2-Hydroxyglutaric aciduria.
Brain researchEnantioseparation of d,l-2-hydroxyglutaric acid by capillary electrophoresis with tandem mass spectrometry-Fast and efficient tool for d- and l-2-hydroxyglutaracidurias diagnosis.
Journal of chromatography. AChiral separation of 2-hydroxyglutaric acid on cinchonan carbamate based weak chiral anion exchangers by high-performance liquid chromatography.
Journal of chromatography. AWhite matter abnormalities in an adult patient with l-2-hydroxyglutaric aciduria.
Brain & developmentIn vivo intracerebral administration of L-2-hydroxyglutaric acid provokes oxidative stress and histopathological alterations in striatum and cerebellum of adolescent rats.
Free radical biology & medicineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Acidúria 2-hidroxiglutárica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Acidúria 2-hidroxiglutárica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- [A case of L-2-hydroxyglutaric aciduria diagnosed with involuntary movements, in which improvement in motor symptoms was achieved following treatment].
- Successful Treatment of 2-hydroxyglutaric Aciduria Diagnosed in Adulthood, Three Decades After the Onset: A Case Report and Literature Review.
- A Pathogenic L2HGDH Variant Impairs Mitochondrial Targeting and Enzyme Function in L-2-Hydroxyglutaric Aciduria: Clinical and Functional Evidence from Two Affected Siblings.
- Genetic, neuroimaging, and clinical characteristics of a cohort of individuals with L-2-hydroxyglutaric aciduria from Türkiye.
- Riboflavin treatment in L-2-hydroxyglutaric aciduria: report on a pediatric patient and literature review.
- Subtle clinical and radiological findings as potential indicators of CNS tumors in L-2-hydroxyglutaric aciduria: a literature review.
- L-2-Hydroxyglutaric Aciduria Complicated by Cerebral Neoplasm.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:19(Orphanet)
- MONDO:0016001(MONDO)
- GARD:10761(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4596888(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
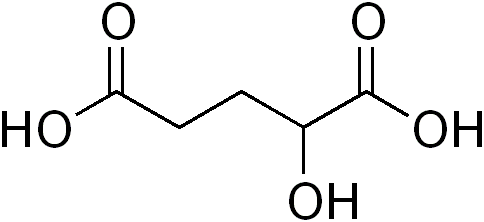
Acidúria 2-hidroxiglutárica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov