Uma doença que tem sua origem em uma alteração na glicosilação O-ligada de proteínas, que é o processo de adicionar açúcares a elas de uma forma específica.
Introdução
O que você precisa saber de cara
Uma doença que tem sua origem em uma alteração na glicosilação O-ligada de proteínas, que é o processo de adicionar açúcares a elas de uma forma específica.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
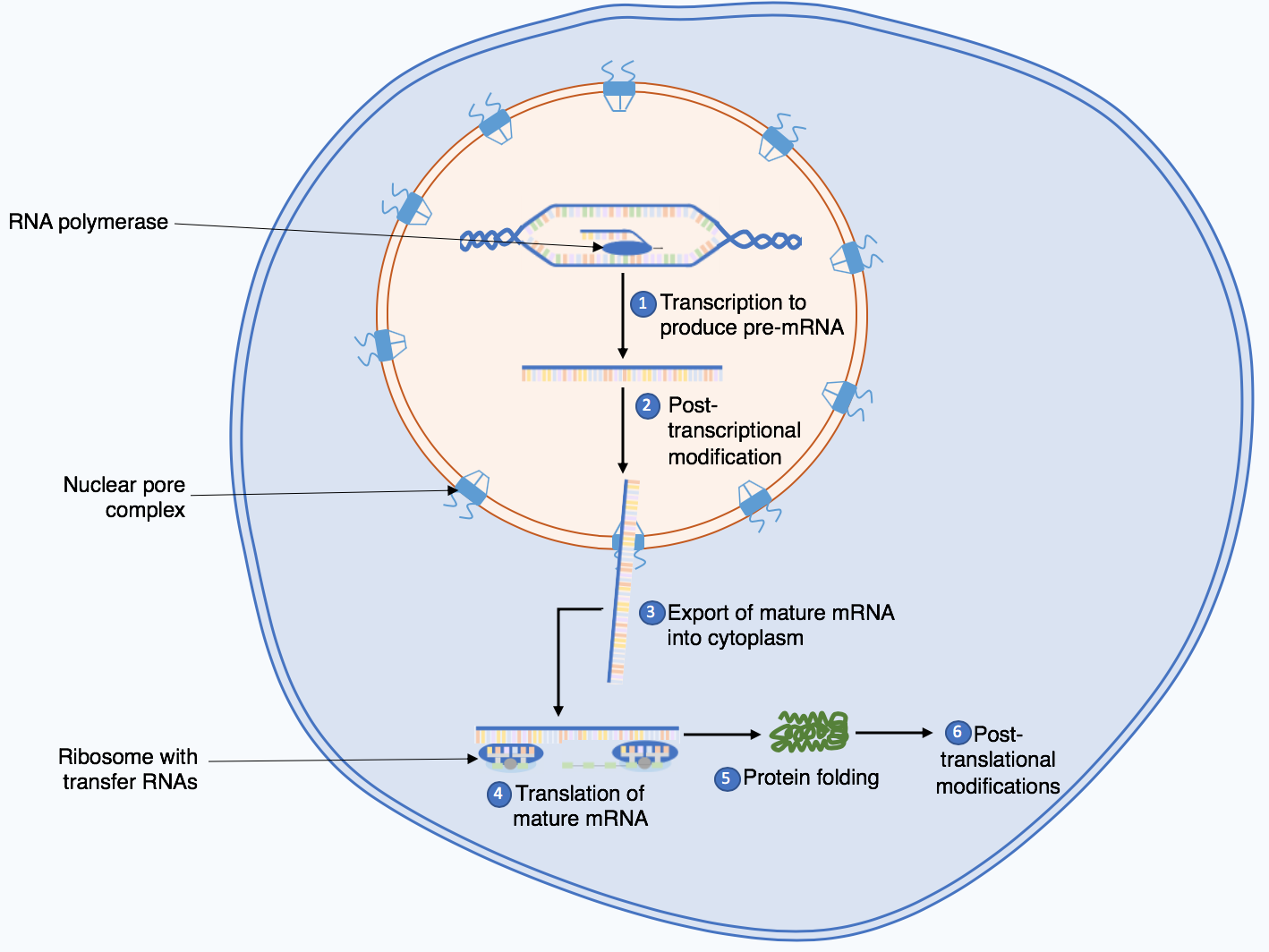
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 157 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 430 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
20 genes identificados com associação a esta condição.
Dual specificity glycosyltransferase that catalyzes the transfer of glucose and xylose from UDP-glucose and UDP-xylose, respectively, to a serine residue found in the consensus sequence of C-X-S-X-P-C (PubMed:21081508, PubMed:21490058, PubMed:21949356, PubMed:27807076, PubMed:28775322). Specifically targets extracellular EGF repeats of protein such as CRB2, F7, F9 and NOTCH2 (PubMed:21081508, PubMed:21490058, PubMed:21949356, PubMed:27807076, PubMed:28775322). Acts as a positive regulator of Not
Endoplasmic reticulum lumen
Dowling-Degos disease 4
A form of Dowling-Degos disease, a genodermatosis manifesting with postpubertal reticulate hyperpigmentation that is progressive and disfiguring, and small hyperkeratotic dark brown papules that affect mainly the flexures and great skin folds. Patients usually show no abnormalities of the hair or nails. DDD4 is characterized by prominent involvement of non-flexural skin areas.
T-box transcription factor that plays an essential role in the determination of the fate of axial stem cells: neural vs mesodermal. Acts in part by down-regulating, a specific enhancer (N1) of SOX2, to inhibit neural development. Seems to play also an essential role in left/right axis determination and acts through effects on Notch signaling around the node as well as through an effect on the morphology and motility of the nodal cilia (By similarity)
Nucleus
Spondylocostal dysostosis 5
A rare condition of variable severity characterized by vertebral and costal anomalies. The main feature include dwarfism, vertebral fusion, hemivertebrae, posterior rib fusion, reduced rib number, and other rib malformations. SCDO5 inheritance can be autosomal dominant or recessive.
Cytidylyltransferase required for protein O-linked mannosylation (PubMed:22522420, PubMed:22522421, PubMed:26687144, PubMed:26923585, PubMed:27130732, PubMed:27601598). Catalyzes the formation of CDP-ribitol nucleotide sugar from D-ribitol 5-phosphate (PubMed:26687144, PubMed:26923585, PubMed:27130732). CDP-ribitol is a substrate of FKTN during the biosynthesis of the phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine-beta-3-N-acetylglucosamine-beta-4-(phosphate-6-)mannose), a carboh
Cytoplasm, cytosol
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A7
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with cobblestone lissencephaly and other brain anomalies, eye malformations, profound intellectual disability, and death usually in the first years of life. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease.
Required for the formation of keratin intermediate filaments in the basal epidermis and maintenance of the skin barrier in response to mechanical stress (By similarity). Regulates the recruitment of Langerhans cells to the epidermis, potentially by modulation of the abundance of macrophage chemotactic cytokines, macrophage inflammatory cytokines and CTNND1 localization in keratinocytes (By similarity)
Cytoplasm
Epidermolysis bullosa simplex 2A, generalized severe
A form of epidermolysis bullosa simplex, a group of skin fragility disorders characterized by skin blistering due to cleavage within the basal layer of keratinocytes, and erosions caused by minor mechanical trauma. There is a broad spectrum of clinical severity ranging from minor blistering on the feet, to subtypes with extracutaneous involvement and a lethal outcome. EBS2A is an autosomal dominant, severe form characterized by extensive intraepidermal blistering from the time of birth with herpetiform marginal spreading and central healing. Oral mucosal involvement, nail dystrophy, onychogryposis, formation of milia, and palmoplantar hyperkeratosis are common features.
Essential subunit of the gamma-secretase complex, an endoprotease complex that catalyzes the intramembrane cleavage of integral membrane proteins such as Notch receptors and APP (amyloid-beta precursor protein) (PubMed:12522139, PubMed:12679784, PubMed:12740439, PubMed:12763021, PubMed:24941111, PubMed:30598546, PubMed:30630874). The gamma-secretase complex plays a role in Notch and Wnt signaling cascades and regulation of downstream processes via its role in processing key regulatory proteins,
Endoplasmic reticulum membraneGolgi apparatus, Golgi stack membraneCell membraneMembrane
Acne inversa, familial, 2, with or without Dowling-Degos disease
An autosomal dominant form of acne inversa, a chronic relapsing inflammatory disease of the hair follicles characterized by recurrent draining sinuses, painful skin abscesses, and disfiguring scars. Manifestations typically appear after puberty. Some ACNINV2 patients also exhibit reticulate hyperpigmentation consistent with Dowling-Degos disease.
Catalyzes the reaction that attaches fucose through an O-glycosidic linkage to a conserved serine or threonine residue found in the consensus sequence C2-X(4,5)-[S/T]-C3 of EGF domains, where C2 and C3 are the second and third conserved cysteines. Specifically uses GDP-fucose as donor substrate and proper disulfide pairing of the substrate EGF domains is required for fucose transfer. Plays a crucial role in NOTCH signaling. Initial fucosylation of NOTCH by POFUT1 generates a substrate for FRINGE
Endoplasmic reticulum
Dowling-Degos disease 2
An autosomal dominant genodermatosis. Affected individuals develop a postpubertal reticulate hyperpigmentation that is progressive and disfiguring, and small hyperkeratotic dark brown papules that affect mainly the flexures and great skin folds. Patients usually show no abnormalities of the hair or nails.
O-linked mannose beta-1,4-N-acetylglucosaminyltransferase that transfers UDP-N-acetyl-D-glucosamine to the 4-position of the mannose to generate N-acetyl-D-glucosamine-beta-1,4-O-D-mannosylprotein. Involved in the biosynthesis of the phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine-beta-3-N-acetylglucosamine-beta-4-(phosphate-6-)mannose), a carbohydrate structure present in alpha-dystroglycan (DAG1), which is required for binding laminin G-like domain-containing extracellular prote
Endoplasmic reticulum membrane
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A8
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with cobblestone lissencephaly and other brain anomalies, eye malformations, profound intellectual disability, and death usually in the first years of life. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease.
Glycosyltransferase that initiates the elongation of O-linked fucose residues attached to EGF-like repeats in the extracellular domain of Notch molecules. Modulates NOTCH1 activity by modifying O-fucose residues at specific EGF-like domains resulting in inhibition of NOTCH1 activation by JAG1 and enhancement of NOTCH1 activation by DLL1 via an increase in its binding to DLL1 (By similarity). Decreases the binding of JAG1 to NOTCH2 but not that of DLL1 (PubMed:11346656). Essential mediator of som
Golgi apparatusGolgi apparatus membrane
Spondylocostal dysostosis 3, autosomal recessive
A condition of variable severity associated with vertebral and rib segmentation defects. The main skeletal malformations include fusion of vertebrae, hemivertebrae, fusion of certain ribs, and other rib malformations. Deformity of the chest and spine (severe scoliosis, kyphoscoliosis and lordosis) is a natural consequence of the malformation and leads to a dwarf-like appearance. As the thorax is small, infants frequently have respiratory insufficiency and repeated respiratory infections resulting in life-threatening complications in the first year of life.
Inhibits primary neurogenesis. May be required to divert neurons along a specific differentiation pathway. Plays a role in the formation of somite boundaries during segmentation of the paraxial mesoderm (By similarity)
Membrane
Spondylocostal dysostosis 1, autosomal recessive
A condition of variable severity associated with vertebral and rib segmentation defects. The main skeletal malformations include fusion of vertebrae, hemivertebrae, fusion of certain ribs, and other rib malformations. Deformity of the chest and spine (severe scoliosis, kyphoscoliosis and lordosis) is a natural consequence of the malformation and leads to a dwarf-like appearance. As the thorax is small, infants frequently have respiratory insufficiency and repeated respiratory infections resulting in life-threatening complications in the first year of life.
Transcriptional repressor. Represses transcription from both N box- and E box-containing promoters. May with HES1, cooperatively regulate somite formation in the presomitic mesoderm (PSM). May function as a segmentation clock, which is essential for coordinated somite segmentation (By similarity)
Nucleus
Spondylocostal dysostosis 4, autosomal recessive
A rare condition of variable severity characterized by vertebral and costal anomalies. The main feature include dwarfism, vertebral fusion, hemivertebrae, posterior rib fusion, reduced rib number, and other rib malformations.
Transfers mannosyl residues to the hydroxyl group of serine or threonine residues. Coexpression of both POMT1 and POMT2 is necessary for enzyme activity, expression of either POMT1 or POMT2 alone is insufficient (PubMed:12369018, PubMed:14699049, PubMed:28512129). Essentially dedicated to O-mannosylation of alpha-DAG1 and few other proteins but not of cadherins and protocaherins (PubMed:28512129)
Endoplasmic reticulum membrane
Muscular dystrophy-dystroglycanopathy congenital with impaired intellectual development B1
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with intellectual disability and mild structural brain abnormalities.
Catalytic subunit of the GMPPA-GMPPB mannose-1-phosphate guanylyltransferase complex (PubMed:33986552). Catalyzes the formation of GDP-mannose, an essential precursor of glycan moieties of glycoproteins and glycolipids (PubMed:33986552). Can catalyze the reverse reaction in vitro (PubMed:33986552). Together with GMPPA regulates GDP-alpha-D-mannose levels (PubMed:33986552)
Cytoplasm
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A14
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with brain anomalies, eye malformations, and profound intellectual disability. The disorder includes a severe form designated as Walker-Warburg syndrome and a less severe phenotype known as muscle-eye-brain disease. MDDGA14 features include increased muscle tone, microcephaly, cleft palate, feeding difficulties, severe muscle weakness, sensorineural hearing loss, cerebellar hypoplasia, ataxia, and retinal dysfunction.
Protein O-mannose kinase that specifically mediates phosphorylation at the 6-position of an O-mannose of the trisaccharide (N-acetylgalactosamine (GalNAc)-beta-1,3-N-acetylglucosamine (GlcNAc)-beta-1,4-mannose) to generate phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine-beta-1,3-N-acetylglucosamine-beta-1,4-(phosphate-6-)mannose). Phosphorylated O-mannosyl trisaccharide is a carbohydrate structure present in alpha-dystroglycan (DAG1), which is required for binding laminin G-like d
Endoplasmic reticulum membrane
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A12
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with cobblestone lissencephaly and other brain anomalies, eye malformations, profound intellectual disability, and death usually in the first years of life. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease.
Transcription factor with important role in somitogenesis. Defines the rostrocaudal patterning of the somite by participating in distinct Notch pathways. Also regulates the FGF signaling pathway. Specifies the rostral half of the somites. Generates rostro-caudal polarity of somites by down-regulating in the presumptive rostral domain DLL1, a Notch ligand. Participates in the segment border formation by activating in the anterior presomitic mesoderm LFNG, a negative regulator of DLL1-Notch signal
Nucleus
Spondylocostal dysostosis 2, autosomal recessive
A condition of variable severity associated with vertebral and rib segmentation defects. The main skeletal malformations include fusion of vertebrae, hemivertebrae, fusion of certain ribs, and other rib malformations. Deformity of the chest and spine (severe scoliosis, kyphoscoliosis and lordosis) is a natural consequence of the malformation and leads to a dwarf-like appearance. As the thorax is small, infants frequently have respiratory insufficiency and repeated respiratory infections resulting in life-threatening complications in the first year of life.
Beta-1,3-glucosyltransferase involved in one of the two pathways responsible for protein O-linked fucosylation, a unique post-translational modification of cysteine-knotted proteins that regulates various biological processes. This pathway targets proteins with Thrombospondin type-1 (TSP1) repeats (TSR) in the endoplasmic reticulum. It starts with POFUT2, which attaches fucose via an O-glycosidic bond to a conserved serine or threonine residue. B3GLCT extends this modification by transferring a
Endoplasmic reticulum membrane
Peters-plus syndrome
An autosomal recessive disorder characterized by anterior eye-chamber abnormalities, disproportionate short stature, developmental delay, characteristic craniofacial features, cleft lip and/or palate.
Transfers mannosyl residues to the hydroxyl group of serine or threonine residues. Coexpression of both POMT1 and POMT2 is necessary for enzyme activity, expression of either POMT1 or POMT2 alone is insufficient (PubMed:14699049, PubMed:28512129). Essentially dedicated to O-mannosylation of alpha-DAG1 and few other proteins but not of cadherins and protocaherins (PubMed:28512129)
Endoplasmic reticulum membrane
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A2
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with cobblestone lissencephaly and other brain anomalies, eye malformations, profound intellectual disability, and death usually in the first years of life. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease.
Catalyzes the transfer of a ribitol-phosphate from CDP-ribitol to the distal N-acetylgalactosamine of the phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine-beta-3-N-acetylglucosamine-beta-4-(phosphate-6-)mannose), a carbohydrate structure present in alpha-dystroglycan (DAG1) (PubMed:26923585, PubMed:27194101, PubMed:29477842). This constitutes the first step in the formation of the ribitol 5-phosphate tandem repeat which links the phosphorylated O-mannosyl trisaccharide to the ligan
Golgi apparatus membraneCytoplasmNucleus
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A4
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with cobblestone lissencephaly and other brain anomalies, eye malformations, profound intellectual disability, and death usually in the first years of life. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease.
Plays a role in somitogenesis. Required for somite segregation and establishment of rostrocaudal polarity in somites (By similarity)
Nucleus
Spondylocostal dysostosis 6, autosomal recessive
A form of spondylocostal dysostosis, a condition of variable severity associated with vertebral and rib segmentation defects. The main skeletal malformations include fusion of vertebrae, hemivertebrae, fusion of certain ribs, and other rib malformations. Deformity of the chest and spine (severe scoliosis, kyphoscoliosis and lordosis) is a natural consequence of the malformation and leads to a dwarf-like appearance. As the thorax is small, infants frequently have respiratory insufficiency and repeated respiratory infections resulting in life-threatening complications in the first year of life.
Catalyzes the transfer of a ribitol 5-phosphate from CDP-L-ribitol to the ribitol 5-phosphate previously attached by FKTN/fukutin to the phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine-beta-3-N-acetylglucosamine-beta-4-(phosphate-6-)mannose), a carbohydrate structure present in alpha-dystroglycan (DAG1) (PubMed:26923585, PubMed:27194101, PubMed:29477842, PubMed:31949166). This constitutes the second step in the formation of the ribose 5-phosphate tandem repeat which links the phos
Golgi apparatus membraneSecretedCell membrane, sarcolemmaRough endoplasmic reticulumCytoplasm
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A5
An autosomal recessive disorder characterized by congenital muscular dystrophy associated with cobblestone lissencephaly and other brain anomalies, eye malformations, profound intellectual disability, and death usually in the first years of life. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease.
Participates in O-mannosyl glycosylation by catalyzing the addition of N-acetylglucosamine to O-linked mannose on glycoproteins (PubMed:11709191, PubMed:27493216, PubMed:28512129). Catalyzes the synthesis of the GlcNAc(beta1-2)Man(alpha1-)O-Ser/Thr moiety on alpha-dystroglycan and other O-mannosylated proteins, providing the necessary basis for the addition of further carbohydrate moieties (PubMed:11709191, PubMed:27493216). Is specific for alpha linked terminal mannose and does not have MGAT3,
Golgi apparatus membrane
Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A3
An autosomal recessive disorder characterized by congenital muscular dystrophy, ocular abnormalities, cobblestone lissencephaly, and cerebellar and pontine hypoplasia. Patients present severe congenital myopia, congenital glaucoma, pallor of the optic disks, retinal hypoplasia, intellectual disability, hydrocephalus, abnormal electroencephalograms, generalized muscle weakness and myoclonic jerks. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease.
Variantes genéticas (ClinVar)
564 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
38 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Alteração da proteína O-glicosilação
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
GALNT3 Inhibits the Progression of Cerebral Ischemia-Reperfusion Injury by Stabilizing TREM2 via O-GalNAc Glycosylation.
A lesão por isquemia-reperfusão cerebral (LIRC) após um AVC é um desafio sem terapias eficazes. Este estudo demonstrou que a enzima GALNT3, cuja expressão diminui na LIRC, pode reduzir significativamente o volume do infarto e melhorar a função neurológica em modelos animais, ao diminuir a inflamação e a morte celular. Isso ocorre porque a GALNT3 estabiliza a proteína TREM2 através de um processo de glicosilação, sugerindo que a manipulação dessa via pode ser uma nova estratégia terapêutica promissora para proteger o cérebro de pacientes com AVC.
🇧🇷 traduzidoGalNAc-T13 maintains neurite architecture and memory retention via O-GalNAc glycosylation of seizure protein 6.
Este estudo demonstra que a enzima GalNAc-T13 é crucial para a formação e manutenção da arquitetura dos neurônios no cérebro (neurite e ramificação dendrítica) e para a consolidação da memória. Sua disfunção, observada em modelos de camundongos, leva a problemas na estrutura cerebral e déficits de memória, que em pacientes poderiam se manifestar como dificuldades cognitivas ou atrasos no desenvolvimento. A GalNAc-T13 age modificando (glicosilando) a proteína SEZ6, processo essencial para a estabilidade e função desta no crescimento neuronal. Compreender este mecanismo oferece insights importantes para médicos e pesquisadores sobre a base molecular de certos distúrbios neuropsiquiátricos e aponta para possíveis alvos diagnósticos e terapêuticos futuros.
🇧🇷 traduzidoProbing APP Cleavage and Amyloid-β Assembly via Synthetic MUC-Type O-Glycosylated APP Glycopeptides.
A glicosilação (adição de açúcares) na proteína precursora amiloide (APP) desempenha um papel fundamental na doença de Alzheimer, influenciando como a APP é processada e como os peptídeos amiloides tóxicos se agregam no cérebro. Este estudo revela que a localização e a quantidade desses açúcares O-GalNAc na APP podem tanto promover a formação de agregados amiloides prejudiciais (especialmente em mutações ligadas à doença), quanto, em outras configurações, desorganizá-los. Compreender esses mecanismos oferece novas perspectivas para o desenvolvimento de diagnósticos e terapias focadas nessas modificações de açúcar para combater a doença.
🇧🇷 traduzidoInterferon-stimulated gene GALNT2 restricts respiratory virus infections.
Este estudo identifica o gene GALNT2, responsável pela O-glicosilação de proteínas, como um importante fator antiviral da imunidade inata. Ele restringe a replicação de vírus respiratórios como coronavírus e influenza, contribuindo para a redução da carga viral e da gravidade da doença. Para pacientes e médicos, é crucial saber que variantes genéticas do GALNT2 com perda de função antiviral foram associadas a um risco elevado de hospitalização após infecção por SARS-CoV-2, destacando a importância da função de O-glicosilação mediada por GALNT2 na defesa contra essas infecções.
🇧🇷 traduzidoDisrupted O-GalNAc glycosylation as a mechanism and biomarker of SLC35A2 -associated epilepsy.
Este estudo revela que a epilepsia grave e os distúrbios neurológicos causados por alterações genéticas no gene SLC35A2 resultam de um defeito específico na produção de um tipo de cadeia de açúcar, as glicanas O-GalNAc, essencial para o desenvolvimento e funcionamento dos neurônios. Essa falha na formação desses açúcares compromete a função neuronal, levando à sua hiperexcitabilidade, o que foi confirmado em modelos e em tecidos cerebrais de pacientes. Para pacientes e médicos, essa descoberta é crucial porque estabelece um mecanismo claro para a doença, abrindo caminho para o desenvolvimento de novos biomarcadores para diagnóstico e monitoramento, e terapias direcionadas para a epilepsia associada ao SLC35A2.
🇧🇷 traduzidoPublicações recentes
Ver todas no PubMed📚 EuropePMCmostrando 199
Disrupted O-GalNAc glycosylation as a mechanism and biomarker of SLC35A2 -associated epilepsy.
bioRxiv : the preprint server for biologyFine structural features of polysaccharides and gut microbiota Co-regulate mucin O-glycosylation: Mechanisms and advances.
Carbohydrate polymersGALNT3 Inhibits the Progression of Cerebral Ischemia-Reperfusion Injury by Stabilizing TREM2 via O-GalNAc Glycosylation.
CNS neuroscience & therapeuticsGalNAc-T13 maintains neurite architecture and memory retention via O-GalNAc glycosylation of seizure protein 6.
Proceedings of the National Academy of Sciences of the United States of AmericaGlycoprotein biomarkers for bladder cancer detection.
Clinica chimica acta; international journal of clinical chemistryProbing APP Cleavage and Amyloid-β Assembly via Synthetic MUC-Type O-Glycosylated APP Glycopeptides.
ACS chemical neuroscienceEpigenetic and O-glycosylation regulation of p66Shc mitigates mitochondrial oxidative stress in aortic dissection.
TheranosticsClinical, biochemical and genetic characterization of an Egyptian patient with SRD5A3-congenital glycosylation disorder.
Ophthalmic geneticsMulti-omics analyses reveal the pathogenic role of terminal ileum-derived IgA+β7+ cells in IgA nephropathy.
Kidney internationalEmergence and epidemiology of dominant variants of human metapneumovirus in the United States between 2016 and 2021.
mBioUnlocking the Sugar Code: Implications and Consequences of Glycosylation in Alzheimer's Disease and Other Tauopathies.
BiomedicinesGALNT7 promotes hepatocellular carcinoma progression by activating the PI3K/AKT signaling pathway via O-glycosylation of MUC13.
Acta biochimica et biophysica SinicaMucin-type O-glycans regulate proteoglycan stability and chondrocyte maturation.
bioRxiv : the preprint server for biologyDynamic glycosylation remodeling in neurological disorders.
Frontiers in molecular neuroscienceGALNT3 is a novel target driving lymphomagenesis via O-glycosylation of FGFR2.
Cell communication and signaling : CCSInterferon-stimulated gene GALNT2 restricts respiratory virus infections.
Nature microbiologyComparative immunoinformatic analysis of Rhipicephalus microplus cocktail vaccine targets.
Parasites & vectorsInsights Into the Pathological Glycosylation Associated With COG6-CDG.
Human mutationα-O-Glycosylation at Tyrosine 10 Promotes the Astrocyte Clearance of Amyloid-β Peptide 1-42.
Chembiochem : a European journal of chemical biologyDecoding the complex substrate specificities of GalNAc-Ts.
GlycobiologyIn vitro cell model to dilucidate the underlying molecular mechanism associated with ophthalmic manifestation of congenital disorders of glycosylation: studying an ALG2-CDG patient.
Frontiers in geneticsPotential Function of Glycosylated RNA in Diseases.
Wiley interdisciplinary reviews. RNAGlycan Signatures on Neutrophils in an Equine Model for Autoimmune Uveitis.
BiomoleculesATP6AP2-Related Disease Caused by Splicing Defects: Abnormal Glycosylation and the First Affected Female.
Journal of inherited metabolic diseaseMolecular dynamics study of the impact of glycosylation on conformational properties of trimeric N-terminal domain of human copper transporter 1.
Carbohydrate researchThe Importance of N- and O-Glycosylation of Brain Cell Surface Glycoproteins.
GlycobiologyMIC13 of Toxoplasma gondii: Potential Gene for Vaccine Candidate-An In Silico Approach.
Journal of parasitology researchAbnormal O-glycan sialylation in the mPFC contributes to depressive-like behaviors in male mice.
Science advancesNeuromuscular Defects in a Drosophila Model of the Congenital Disorder of Glycosylation SLC35A2-CDG.
BiomoleculesSalivary Nitrate Maintains Mucosal Homeostasis via the Sialin-Neuropeptide Axis.
Journal of dental researchThe β-1,4 GalT-V Interactome-Potential Therapeutic Targets and a Network of Pathways Driving Cancer and Cardiovascular and Inflammatory Diseases.
International journal of molecular sciencesProtein glycosylation modification as a novel forensic biomarker for discriminating monozygotic twins.
Forensic science internationalHylocereus polyrhizus Pulp Residues Polysaccharide Alleviates High-Fat Diet-Induced Obesity by Modulating Intestinal Mucus Secretion and Glycosylation.
Foods (Basel, Switzerland)Is dolichol pathway dysfunction a significant factor in Alzheimer's disease?
InflammopharmacologyIntegrated multi-omics analysis and machine learning refine molecular subtypes and prognosis in hepatocellular carcinoma through O-linked glycosylation genes.
Functional & integrative genomicsApproaches to diagnostic screening for congenital disorders of glycosylation and its prevalence in Japan.
Journal of human geneticsGlycoIP: an integrated platform for simultaneous and site-specific N/O-glycosylation analysis of human semen.
Frontiers in chemistryMap of the neuronal O-glycoproteome reveals driver functions in the regulated secretory pathway.
The Journal of biological chemistryGALNT14 deficiency: connecting multiple links in the IgA nephropathy pathogenetic chain.
The Journal of clinical investigationImplications of Mucin-Type O-Glycosylation in Alzheimer's Disease.
Molecules (Basel, Switzerland)Molecular mechanisms and pathophysiological implications of mucin-type O-glycosylation dysregulation in colorectal cancer progression.
Naunyn-Schmiedeberg's archives of pharmacologyMyriad mechanisms: factors regulating the synthesis of aberrant mucin-type O-glycosylation found on cancer cells.
GlycobiologyNew insights into phytochemicals via protein glycosylation focused on aging and diabetes.
Phytomedicine : international journal of phytotherapy and phytopharmacologyGlycosylation in Dermatology: Unveiling the Sugar Coating of Skin Disease.
Experimental dermatologyClinical and Molecular Features of Patients With Congenital Disorders of Glycosylation in Japan.
JIMD reportsLoss of GalNAc-T14 links O-glycosylation defects to alterations in B cell homing in IgA nephropathy.
The Journal of clinical investigationDiagnostic and Therapeutic Approaches in Congenital Disorders of Glycosylation.
Handbook of experimental pharmacologySPOP/NOLC1/B4GALT1 signaling axis enhances paclitaxel resistance in endometrial cancer by inducing O-dysglycosylation.
OncogeneExploring the combined roles of GALNT1 and GALNT2 in hepatocellular carcinoma malignancy and EGFR modulation.
Discover oncologyER O-glycosylation in synovial fibroblasts drives cartilage degradation.
Nature communicationsEmerging Biochemical and Immunologic Mechanisms in the Pathogenesis of IgA Nephropathy.
Seminars in nephrologyRCL glycosylation of serum corticosteroid-binding globulin: implications in cortisol delivery and septic shock.
GlycobiologyAntibodies with specificity to glycan motifs that decorate OMV cargo proteins.
mSphereO-GalNAc glycans are enriched in neuronal tracts and regulate nodes of Ranvier.
Proceedings of the National Academy of Sciences of the United States of AmericaC1GALT1C1-Associated Mosaic Disorder of Glycosylation in a Female.
Journal of inherited metabolic diseaseMass Spectrometry as a First-Line Diagnostic Aid for Congenital Disorders of Glycosylation.
Mass spectrometry (Tokyo, Japan)Exonic CircGUCY1A2 inhibits pulmonary artery smooth muscle cells phenotypic switching via regulating O-glycosylation of COL3A1 in pulmonary hypertension.
European journal of pharmacologyO-GlcNAc modification differentially regulates microtubule binding and pathological conformations of tau isoforms in vitro.
The Journal of biological chemistryEnhanced O-glycosylation site prediction using explainable machine learning technique with spatial local environment.
Bioinformatics (Oxford, England)Brain microenvironment-remodeling nanomedicine improves cerebral glucose metabolism, mitochondrial activity and synaptic function in a mouse model of Alzheimer's disease.
BiomaterialsGlycosylation Pathways Targeted by Deregulated miRNAs in Autism Spectrum Disorder.
International journal of molecular sciencesDecoding Extracellular Protein Glycosylation in Human Health and Disease.
Annual review of analytical chemistry (Palo Alto, Calif.)SLC10A7 regulates O-GalNAc glycosylation and Ca2+ homeostasis in the secretory pathway: insights into SLC10A7-CDG.
Cellular and molecular life sciences : CMLSGolgi pH elevation due to loss of V-ATPase subunit V0a2 function correlates with tissue-specific glycosylation changes and globozoospermia.
Cellular and molecular life sciences : CMLS"Intrinsic disorder-protein modification-LLPS-tumor" regulatory axis: From regulatory mechanisms to precision medicine.
Biochimica et biophysica acta. Reviews on cancerGalNT2-mediated O-glycosylation affects pancreas development and function in mice.
Scientific reportsGALNT6, transcriptionally inhibited by KLF9, promotes osteosarcoma progression by increasing EFEMP1 expression via O-glycosylation modification.
Biochimica et biophysica acta. Molecular cell researchC1GALT1 high expression enhances the progression of glioblastoma through the EGFR-AKT/ERK cascade.
Cellular signallingGALNT3 in Ischemia-Reperfusion Injury of the Kidney.
Journal of the American Society of Nephrology : JASNMALAT1/miR-582-5p/GALNT1/MUC1 axis modulates progression of AML leukemia stem cells by regulating JAK2/STAT3 pathway.
Annals of hematologyGiantin mediates Golgi localization of Gal3-O-sulfotransferases and affects salivary mucin sulfation in patients with Sjögren's disease.
JCI insightRapid identification of primary atopic disorders (PAD) by a clinical landmark-guided, upfront use of genomic sequencing.
Allergologie selectHormonal and Allosteric Regulation of the Luteinizing Hormone/Chorionic Gonadotropin Receptor.
Frontiers in bioscience (Landmark edition)Aberrant glycosylation in schizophrenia: insights into pathophysiological mechanisms and therapeutic potentials.
Frontiers in pharmacologyO-Glycosylation of a male seminal fluid protein influences sperm binding and female postmating behavior.
PNAS nexusClinical glycoprotein mass spectrometry: The future of disease detection and monitoring.
Journal of mass spectrometry : JMSThe genetics and epidemiology of N- and O-immunoglobulin A glycomics.
Genome medicineIn vivo mapping of the mouse Galnt3-specific O-glycoproteome.
The Journal of biological chemistryO-glycosylation of IgA1 and the pathogenesis of an autoimmune disease IgA nephropathy.
GlycobiologySite-Specific Glycosylation Analysis of Human and Murine Fcγ Receptor II Family Members Reveals Variant-Specific N-Glycosylation.
Journal of proteome researchB3galt5 functions as a PXR target gene and regulates obesity and insulin resistance by maintaining intestinal integrity.
Nature communicationsInsights into molecular and cellular functions of the Golgi calcium/manganese-proton antiporter TMEM165.
The Journal of biological chemistryComputational design of novel chimeric multiepitope vaccine against bacterial and viral disease in tilapia (Oreochromis sp.).
Scientific reportsB3GNT7 regulates mucin O-glycosylation to alleviate colonic inflammation.
BMC gastroenterologyThe intriguing strategies of Tannerella forsythia's host interaction.
Frontiers in oral healthApolipoprotein-CIII O-Glycosylation Is Associated with Micro- and Macrovascular Complications of Type 2 Diabetes.
International journal of molecular sciencesMatriglycan maintains t-tubule structural integrity in cardiac muscle.
Proceedings of the National Academy of Sciences of the United States of AmericaDiaminocyclopentane - l-Lysine Adducts: Potent and selective inhibitors of human O-GlcNAcase.
Bioorganic chemistryBiochemical diagnosis of congenital disorders of glycosylation.
Advances in clinical chemistryComprehensive Glycosylation Characterization of Recombinant Human Erythropoietin by Electron-Activated Dissociation Mass Spectrometry.
Applied biochemistry and biotechnologyA Toxoplasma gondii O-glycosyltransferase that modulates bradyzoite cyst wall rigidity is distinct from host homologues.
Nature communicationsThe importance of muscle glycogen phosphorylase in glial cells function.
Biochemical Society transactionsGenomic analysis of canine pneumoviruses and canine respiratory coronavirus from New Zealand.
New Zealand veterinary journalO-glycan determinants regulate VWF trafficking to Weibel-Palade bodies.
Blood advancesStool Glycoproteomics Signatures of Pre-Cancerous Lesions and Colorectal Cancer.
International journal of molecular sciencesImpaired Glycosylation of Gastric Mucins Drives Gastric Tumorigenesis and Serves as a Novel Therapeutic Target.
GastroenterologyProtein glycosylation in lung cancer from a mass spectrometry perspective.
Mass spectrometry reviewsProfiling Human Cerebrospinal Fluid (CSF) Endogenous Peptidome in Alzheimer's Disease.
Methods in molecular biology (Clifton, N.J.)Aberrantly Glycosylated GLUT1 as a Poor Prognosis Marker in Aggressive Bladder Cancer.
International journal of molecular sciencesIon mobility-tandem mass spectrometry of mucin-type O-glycans.
Nature communicationsEmerging Roles of UDP-GalNAc Polypeptide N-Acetylgalactosaminyltransferases in Cardiovascular Disease.
Aging and diseaseFunctional prediction of proteins from the human gut archaeome.
ISME communicationsLoss of the glycosyltransferase Galnt11 affects vitamin D homeostasis and bone composition.
The Journal of biological chemistryExamining the association between serum galactose-deficient IgA1 and primary IgA nephropathy: a systematic review and meta-analysis.
Journal of nephrologyDeficiency of polypeptide N-acetylgalactosamine transferase 9 contributes to a risk for Parkinson's disease via mitochondrial dysfunctions.
International journal of biological macromoleculesHSF1 Increases EOGT-Mediated Glycosylation of Notch1 to Promote IL-1β-Induced Inflammatory Injury of Chondrocytes.
CartilageCharacterization of MdpS: an in-depth analysis of a MUC5B-degrading protease from Streptococcus oralis.
Frontiers in microbiologyO-glycoprofiling of Serum Apolipoprotein C-III in Colorectal Cancer.
Frontiers in bioscience (Landmark edition)Fecal-adherent mucus is a non-invasive source of primary human MUC2 for structural and functional characterization in health and disease.
The Journal of biological chemistryTRIM14 suppressed the progression of NSCLC via hexosamine biosynthesis pathway.
CarcinogenesisCancer snap-shots: Biochemistry and glycopathology of O-glycans: A review.
International journal of biological macromoleculesProtein O-GlcNAcylation: The sweet hub in liver metabolic flexibility from a (patho)physiological perspective.
Liver international : official journal of the International Association for the Study of the LiverAdvances in Bacterial Oligosaccharyltransferase Structure Elucidation and Potential Application to Glycoconjugate Vaccine Design.
Frontiers in bioscience (Landmark edition)H3K27 acetylation activated-PDLIM7 promotes castration-resistant prostate cancer progression by inducing O-Glycosylation of YAP1 protein.
Translational oncologyTalniflumate abrogates mucin immune suppressive barrier improving efficacy of gemcitabine and nab-paclitaxel treatment in pancreatic cancer.
Journal of translational medicineImpaired digestive function of sucrase-isomaltase in a complex with the Greenlandic sucrase-isomaltase variant.
Biochimica et biophysica acta. Molecular basis of diseaseGlycoTCFM: Glycoproteomics Based on Two Complementary Fragmentation Methods Reveals Distinctive O-Glycosylation in Human Sperm and Seminal Plasma.
Journal of proteome researchMuc2 mucin O-glycosylation interacts with enteropathogenic Escherichia coli to influence the development of ulcerative colitis based on the NF-kB signaling pathway.
Journal of translational medicineQuantitative mapping of the in vivo O-GalNAc glycoproteome in mouse tissues identifies GalNAc-T2 O-glycosites in metabolic disorder.
Proceedings of the National Academy of Sciences of the United States of AmericaApolipoprotein-CIII O-Glycosylation, a Link between GALNT2 and Plasma Lipids.
International journal of molecular sciencesExploiting Chemical Protein Synthesis to Study the Role of Tyrosine Sulfation on Anticoagulants from Hematophagous Organisms.
Accounts of chemical researchProteomic analysis of diabetic retinas.
Frontiers in endocrinologyHighly functionalized diaminocyclopentanes: A new route to potent and selective inhibitors of human O-GlcNAcase.
Bioorganic chemistryProtein glycosylation: bridging maternal-fetal crosstalk during embryo implantation†.
Biology of reproductionPolypeptide N-acetylgalactosaminyltransferase (GalNAc-T) isozyme surface charge governs charge substrate preferences to modulate mucin type O-glycosylation.
GlycobiologyIn Silico Characterization of an Important Metacyclogenesis Marker in Leishmania donovani, HASPB1, as a Potential Vaccine Candidate.
BioMed research internationalDeciphering Protein O-GalNAcylation: Method Development and Disease Implication.
ACS omegaO-GalNAc glycosylation determines intracellular trafficking of APP and Aβ production.
The Journal of biological chemistryGermline C1GALT1C1 mutation causes a multisystem chaperonopathy.
Proceedings of the National Academy of Sciences of the United States of AmericaGlycosylation and behavioral symptoms in neurological disorders.
Translational psychiatryEmerging roles of O-glycosylation in regulating protein aggregation, phase separation, and functions.
Current opinion in chemical biologyAltered Glycosylation in Progression and Management of Bladder Cancer.
Molecules (Basel, Switzerland)Clinical Presentation of a Patient with a Congenital Disorder of Glycosylation, Type IIs (ATP6AP1), and Liver Transplantation.
International journal of molecular sciencesApolipoprotein E O-glycosylation is associated with amyloid plaques and APOE genotype.
Analytical biochemistryBiochemical characterization, and anti-inflammatory and antitumor activities of glycoprotein from lamb abomasum.
Journal of ethnopharmacologyIndoxyl sulfate induces left ventricular hypertrophy via the AhR-FGF23-FGFR4 signaling pathway.
Frontiers in cardiovascular medicineIdentification of global inhibitors of cellular glycosylation.
Nature communicationsIn-depth quantitative proteomics analysis revealed C1GALT1 depletion in ECC-1 cells mimics an aggressive endometrial cancer phenotype observed in cancer patients with low C1GALT1 expression.
Cellular oncology (Dordrecht, Netherlands)The (Sialyl) Tn antigen: Contributions to immunosuppression in gastrointestinal cancers.
Frontiers in oncologyCase report: The art of anesthesiology-Approaching a minor procedure in a child with MPI-CDG.
Frontiers in pharmacologyEmerging Roles of the Unique Molecular Chaperone Cosmc in the Regulation of Health and Disease.
BiomoleculesGALNT1 Enhances Malignant Phenotype of Gastric Cancer via Modulating CD44 Glycosylation to Activate the Wnt/β-catenin Signaling Pathway.
International journal of biological sciencesSimultaneous Quantification of Apolipoprotein C-III O-Glycoforms by Protein-MRM.
Journal of proteome researchDevelopment of an enrichment-free one-pot sample preparation and ultra-high performance liquid chromatography-tandem mass spectrometry method to identify Immunoglobulin A1 hinge region O-glycoforms for Immunoglobulin A nephropathy.
Journal of chromatography. AA State of Natriuretic Peptide Deficiency.
Endocrine reviewsGlycosylation in SARS-CoV-2 variants: A path to infection and recovery.
Biochemical pharmacologyProtein O-GlcNAcylation in Metabolic Modulation of Skeletal Muscle: A Bright but Long Way to Go.
MetabolitesGlobal mapping of GalNAc-T isoform-specificities and O-glycosylation site-occupancy in a tissue-forming human cell line.
Nature communicationsClinical and molecular findings in three Japanese patients with N-acetylneuraminic acid synthetase-congenital disorder of glycosylation (NANS-CDG).
Scientific reportsA novel FGF23 mutation in hyperphosphatemic familial tumoral calcinosis and its deleterious effect on protein O-glycosylation.
Frontiers in endocrinologyFringe-positive Golgi outposts unite temporal Furin 2 convertase activity and spatial Delta signal to promote dendritic branch retraction.
Cell reportsLysosomal cathepsin D mediates endogenous mucin glycodomain catabolism in mammals.
Proceedings of the National Academy of Sciences of the United States of AmericaO-Glycosylating Enzyme GALNT2 Predicts Worse Prognosis in Cervical Cancer.
Pathology oncology research : PORRecent advances in demystifying O-glycosylation in health and disease.
ProteomicsThe role and potential mechanism of O-Glycosylation in gastrointestinal tumors.
Pharmacological researchHeterogeneous glycosylation and methylation of the Aeromonas caviae flagellin.
MicrobiologyOpenSi-Wu Water Extracts Protect against Colonic Mucus Barrier Damage by Regulating Muc2 Mucin Expression in Mice Fed a High-Fat Diet.
Foods (Basel, Switzerland)O-Glycosylation Changes in Serum Immunoglobulin G Are Associated with Inflammation Development in Advanced Endometriosis.
International journal of molecular sciencesO-Glycan-Dependent Interaction between MUC1 Glycopeptide and MY.1E12 Antibody by NMR, Molecular Dynamics and Docking Simulations.
International journal of molecular sciencesPost-Translational Modifications in Atopic Dermatitis: Current Research and Clinical Relevance.
Frontiers in cell and developmental biologyAdvances in glycopeptide enrichment methods for the analysis of protein glycosylation over the past decade.
Journal of separation scienceGlycosylation of HDL-Associated Proteins and Its Implications in Cardiovascular Disease Diagnosis, Metabolism and Function.
Frontiers in cardiovascular medicineDifferential Effects of D-Galactose Supplementation on Golgi Glycosylation Defects in TMEM165 Deficiency.
Frontiers in cell and developmental biologyMucin-Type O-Glycosylation Proximal to β-Secretase Cleavage Site Affects APP Processing and Aggregation Fate.
Frontiers in chemistryThe barrier and beyond: Roles of intestinal mucus and mucin-type O-glycosylation in resistance and tolerance defense strategies guiding host-microbe symbiosis.
Gut microbesIn-Depth Profiling of O-Glycan Isomers in Human Cells Using C18 Nanoliquid Chromatography-Mass Spectrometry and Glycogenomics.
Analytical chemistryGlobal Loss of Core 1-Derived O-Glycans in Mice Leads to High Mortality Due to Acute Kidney Failure and Gastric Ulcers.
International journal of molecular sciencesPan-cancer analysis of GALNTs expression identifies a prognostic of GALNTs feature in low grade glioma.
Journal of leukocyte biologyCOG6-CDG: Novel variants and novel malformation.
Birth defects researchCharacterization of the O-Glycoproteome of Porphyromonas gingivalis.
Microbiology spectrumGALNT4 primes monocytes adhesion and transmigration by regulating O-Glycosylation of PSGL-1 in atherosclerosis.
Journal of molecular and cellular cardiologyIn-Depth Site-Specific O-Glycosylation Analysis of Glycoproteins and Endogenous Peptides in Cerebrospinal Fluid (CSF) from Healthy Individuals, Mild Cognitive Impairment (MCI), and Alzheimer's Disease (AD) Patients.
ACS chemical biologyAsparagine Tautomerization in Glycosyltransferase Catalysis. The Molecular Mechanism of Protein O-Fucosyltransferase 1.
ACS catalysisO-Glycosylation Induces Amyloid-β To Form New Fibril Polymorphs Vulnerable for Degradation.
Journal of the American Chemical SocietyIntegrated N- and O-Glycomics of Acute Myeloid Leukemia (AML) Cell Lines.
CellsAltered Mucus Barrier Integrity and Increased Susceptibility to Colitis in Mice upon Loss of Telocyte Bone Morphogenetic Protein Signalling.
CellsMucin-Type O-Glycans: Barrier, Microbiota, and Immune Anchors in Inflammatory Bowel Disease.
Journal of inflammation researchO-GlcNAcase inhibitor has protective effects in intracerebral hemorrhage by suppressing the inflammatory response.
NeuroreportImmunoglobulin A Glycosylation and Its Role in Disease.
Experientia supplementum (2012)Lectin and Liquid Chromatography-Based Methods for Immunoglobulin (G) Glycosylation Analysis.
Experientia supplementum (2012)Polypeptide N-acetylgalactosaminyltransferase-Associated Phenotypes in Mammals.
Molecules (Basel, Switzerland)O-Glycosylation Landscapes of SARS-CoV-2 Spike Proteins.
Frontiers in chemistryCharacterization of the O-Glycoproteome of Tannerella forsythia.
mSphereMucolytic bacteria: prevalence in various pathological diseases.
World journal of microbiology & biotechnologyMucin-Type O-GalNAc Glycosylation in Health and Disease.
Advances in experimental medicine and biologyA novel mechanism for C1GALT1 in the regulation of gastric cancer progression.
Cell & bioscienceGlobal functions of O-glycosylation: promises and challenges in O-glycobiology.
The FEBS journalO-glycosylation pattern of the SARS-CoV-2 spike protein reveals an "O-Follow-N" rule.
Cell researchTyrosine O-GalNAc Alters the Conformation and Proteolytic Susceptibility of APP Model Glycopeptides.
ACS chemical neuroscienceA semi-automated, high throughput approach for O-glycosylation profiling of in vitro established cancer cell lines by MALDI-FT-ICR MS.
Glycoconjugate journalComprehensive analysis of O-glycosylation of amyloid precursor protein (APP) using targeted and multi-fragmentation MS strategy.
Biochimica et biophysica acta. General subjectsThe O-Glycome of Human Nigrostriatal Tissue and Its Alteration in Parkinson's Disease.
Journal of proteome researchO-glycoforms of polymeric immunoglobulin A1 in the plasma of patients with IgA nephropathy are associated with pathological phenotypes.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationC1GALT1 in health and disease.
Oncology lettersO- and N-Glycosylation of Serum Immunoglobulin A is Associated with IgA Nephropathy and Glomerular Function.
Journal of the American Society of Nephrology : JASNBAG3 epigenetically regulates GALNT10 expression via WDR5 and facilitates the stem cell-like properties of platin-resistant ovarian cancer cells.
Biochimica et biophysica acta. Molecular cell researchOxonium Ion Guided Analysis of Quantitative Proteomics Data Reveals Site-Specific O-Glycosylation of Anterior Gradient Protein 2 (AGR2).
International journal of molecular sciencesProteoglycan synthesis in conserved oligomeric Golgi subunit deficient HEK293T cells is affected differently, depending on the lacking subunit.
Traffic (Copenhagen, Denmark)Slimy partners: the mucus barrier and gut microbiome in ulcerative colitis.
Experimental & molecular medicineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Alteração da proteína O-glicosilação.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Alteração da proteína O-glicosilação
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- GALNT3 Inhibits the Progression of Cerebral Ischemia-Reperfusion Injury by Stabilizing TREM2 via O-GalNAc Glycosylation.
- GalNAc-T13 maintains neurite architecture and memory retention via O-GalNAc glycosylation of seizure protein 6.Proceedings of the National Academy of Sciences of the United States of America· 2026· PMID 41790942mais citado
- Probing APP Cleavage and Amyloid-β Assembly via Synthetic MUC-Type O-Glycosylated APP Glycopeptides.
- Interferon-stimulated gene GALNT2 restricts respiratory virus infections.
- Disrupted O-GalNAc glycosylation as a mechanism and biomarker of SLC35A2 -associated epilepsy.
- PGM1 deficiency: Substrate use during exercise and effect of treatment with galactose.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:309447(Orphanet)
- MONDO:0017741(MONDO)
- GARD:21336(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55787317(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Alteração da proteína O-glicosilação
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata