A dicarboxilicaminoacidúria é caracterizada por hipoglicemia e hiperprolinemia de início infantil associadas, em certos casos, a déficit intelectual.
Introdução
O que você precisa saber de cara
A dicarboxilicaminoacidúria é caracterizada por hipoglicemia e hiperprolinemia de início infantil associadas, em certos casos, a déficit intelectual.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 3 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 6 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
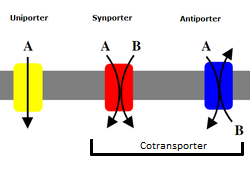
fontes oficiaisSodium-dependent, high-affinity amino acid transporter that mediates the uptake of L-glutamate and also L-aspartate and D-aspartate (PubMed:21123949, PubMed:26690923, PubMed:33658209, PubMed:7521911, PubMed:7914198, PubMed:8857541). Can also transport L-cysteine (PubMed:21123949). Functions as a symporter that transports one amino acid molecule together with two or three Na(+) ions and one proton, in parallel with the counter-transport of one K(+) ion (PubMed:26690923, PubMed:33658209, PubMed:75
Cell membraneApical cell membraneSynapse, synaptosomeEarly endosome membraneLate endosome membraneRecycling endosome membrane
Dicarboxylic aminoaciduria
An autosomal recessive disorder characterized by abnormal excretion of urinary glutamate and aspartate, resulting from the incomplete reabsorption of anionic amino acids from the glomerular filtrate in the kidney. It can be associated with intellectual disability.
Variantes genéticas (ClinVar)
177 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 111 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Aminoacidúria dicarboxílica
Centros de Referência SUS
21 centros habilitados pelo SUS para Aminoacidúria dicarboxílica
Centros para Aminoacidúria dicarboxílica
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
NUPAD / Faculdade de Medicina UFMG
Av. Prof. Alfredo Balena, 189 - 5 andar - Centro, Belo Horizonte - MG, 30130-100 · CNES 2183226
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital de Clínicas da Universidade Federal de Pernambuco
Av. Prof. Moraes Rego, 1235 - Cidade Universitária, Recife - PE, 50670-901 · CNES 2561492
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital Universitário Onofre Lopes (HUOL)
Av. Nilo Peçanha, 620 - Petrópolis, Natal - RN, 59012-300 · CNES 2408570
Atenção Especializada
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Instituto da Criança e do Adolescente (ICr-HCFMUSP)
Av. Dr. Enéas Carvalho de Aguiar, 647 - Cerqueira César, São Paulo - SP, 05403-000 · CNES 2081695
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Aspartic Acid in Health and Disease.
Aspartic acid exists in L- and D-isoforms (L-Asp and D-Asp). Most L-Asp is synthesized by mitochondrial aspartate aminotransferase from oxaloacetate and glutamate acquired by glutamine deamidation, particularly in the liver and tumor cells, and transamination of branched-chain amino acids (BCAAs), particularly in muscles. The main source of D-Asp is the racemization of L-Asp. L-Asp transported via aspartate-glutamate carrier to the cytosol is used in protein and nucleotide synthesis, gluconeogenesis, urea, and purine-nucleotide cycles, and neurotransmission and via the malate-aspartate shuttle maintains NADH delivery to mitochondria and redox balance. L-Asp released from neurons connects with the glutamate-glutamine cycle and ensures glycolysis and ammonia detoxification in astrocytes. D-Asp has a role in brain development and hypothalamus regulation. The hereditary disorders in L-Asp metabolism include citrullinemia, asparagine synthetase deficiency, Canavan disease, and dicarboxylic aminoaciduria. L-Asp plays a role in the pathogenesis of psychiatric and neurologic disorders and alterations in BCAA levels in diabetes and hyperammonemia. Further research is needed to examine the targeting of L-Asp metabolism as a strategy to fight cancer, the use of L-Asp as a dietary supplement, and the risks of increased L-Asp consumption. The role of D-Asp in the brain warrants studies on its therapeutic potential in psychiatric and neurologic disorders.
Amino Acid Transport Across the Mammalian Intestine.
The small intestine mediates the absorption of amino acids after ingestion of protein and sustains the supply of amino acids to all tissues. The small intestine is an important contributor to plasma amino acid homeostasis, while amino acid transport in the large intestine is more relevant for bacterial metabolites and fluid secretion. A number of rare inherited disorders have contributed to the identification of amino acid transporters in epithelial cells of the small intestine, in particular cystinuria, lysinuric protein intolerance, Hartnup disorder, iminoglycinuria, and dicarboxylic aminoaciduria. These are most readily detected by analysis of urine amino acids, but typically also affect intestinal transport. The genes underlying these disorders have all been identified. The remaining transporters were identified through molecular cloning techniques to the extent that a comprehensive portrait of functional cooperation among transporters of intestinal epithelial cells is now available for both the basolateral and apical membranes. Mouse models of most intestinal transporters illustrate their contribution to amino acid homeostasis and systemic physiology. Intestinal amino acid transport activities can vary between species, but these can now be explained as differences of amino acid transporter distribution along the intestine. © 2019 American Physiological Society. Compr Physiol 9:343-373, 2019.
Publicações recentes
Aspartic Acid in Health and Disease.
Amino Acid Transport Across the Mammalian Intestine.
Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria.
Apical transporters for neutral amino acids: physiology and pathophysiology.
Aminoacidurias: Clinical and molecular aspects.
📚 EuropePMC9 artigos no totalmostrando 2
Aspartic Acid in Health and Disease.
NutrientsAmino Acid Transport Across the Mammalian Intestine.
Comprehensive PhysiologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Aminoacidúria dicarboxílica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Aminoacidúria dicarboxílica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Aspartic Acid in Health and Disease.
- Amino Acid Transport Across the Mammalian Intestine.
- Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria.
- Apical transporters for neutral amino acids: physiology and pathophysiology.
- Aminoacidurias: Clinical and molecular aspects.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2195(Orphanet)
- OMIM OMIM:222730(OMIM)
- MONDO:0009110(MONDO)
- GARD:1855(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q16252204(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Aminoacidúria dicarboxílica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata