O dismorfismo digitotalar, também conhecido como artrogripose distal tipo 1 (DA1), é uma anomalia congênita autossômica dominante caracterizada por contraturas das regiões distais das mãos e pés, sem envolvimento facial ou quaisquer anomalias adicionais. É o tipo mais comum de artrogripose distal.
Introdução
O que você precisa saber de cara
O dismorfismo digitotalar, também conhecido como artrogripose distal tipo 1 (DA1), é uma anomalia congênita autossômica dominante caracterizada por contraturas das regiões distais das mãos e pés, sem envolvimento facial ou quaisquer anomalias adicionais. É o tipo mais comum de artrogripose distal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 40 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
6 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Muscle contraction
Cytoplasm, myofibril
Arthrogryposis, distal, 2A
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA2A is characterized by contractures of the hands and feet, oropharyngeal abnormalities, scoliosis, and a distinctive face that includes a very small oral orifice, puckered lips, and a H-shaped dimple of the chin.
Troponin T is the tropomyosin-binding subunit of troponin, the thin filament regulatory complex which confers calcium-sensitivity to striated muscle actomyosin ATPase activity
Arthrogryposis, distal, 2B2
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. Distal arthrogryposis type 2 is characterized by contractures of the hands and feet, and a distinctive face characterized by prominent nasolabial folds, small mouth and downslanting palpebral fissures. DA2B2 inheritance is autosomal dominant.
Troponin I is the inhibitory subunit of troponin, the thin filament regulatory complex which confers calcium-sensitivity to striated muscle actomyosin ATPase activity
Arthrogryposis, distal, 2B1
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA2B is characterized by contractures of the hands and feet, and a distinctive face characterized by prominent nasolabial folds, small mouth and downslanting palpebral fissures. DA2B1 inheritance is autosomal dominant.
Voltage-gated ion channel responsible for the resting Na(+) permeability that controls neuronal excitability (PubMed:17448995, PubMed:31409833). NALCN channel functions as a multi-protein complex, which consists at least of NALCN, NALF1, UNC79 and UNC80 (PubMed:32494638, PubMed:33203861). NALCN is the voltage-sensing, pore-forming subunit of the NALCN channel complex (PubMed:17448995). NALCN channel complex is constitutively active and conducts monovalent cations but is blocked by physiological
Cell membrane
Hypotonia, infantile, with psychomotor retardation and characteristic facies 1
A neurodegenerative disease characterized by variable degrees of hypotonia, speech impairment, intellectual disability, pyramidal signs, subtle facial dysmorphism, and chronic constipation. Some patients manifest neuroaxonal dystrophy, optic atrophy, unmyelinated axons and spheroid bodies in tissue biopsies.
Binds to actin filaments in muscle and non-muscle cells. Plays a central role, in association with the troponin complex, in the calcium dependent regulation of vertebrate striated muscle contraction. Smooth muscle contraction is regulated by interaction with caldesmon. In non-muscle cells is implicated in stabilizing cytoskeleton actin filaments. The non-muscle isoform may have a role in agonist-mediated receptor internalization
Cytoplasm, cytoskeleton
Congenital myopathy 23
An autosomal dominant muscular disorder characterized clinically by hypotonia and muscle weakness, and a static or slowly progressive clinical course. Disease onset ranges from birth to childhood. Histologic examination of muscle fibers shows various anomalies including fiber type disproportion, an irregular myofibrillar network, abnormal thread-like or rod-shaped structures, and cap-like structures which are well demarcated and peripherally located under the sarcolemma with abnormal accumulation of sarcomeric proteins.
Thick filament-associated protein located in the crossbridge region of vertebrate striated muscle a bands. Slow skeletal protein that binds to both myosin and actin (PubMed:31025394, PubMed:31264822). In vitro, binds to native thin filaments and modifies the activity of actin-activated myosin ATPase. May modulate muscle contraction or may play a more structural role
Arthrogryposis, distal, 1B
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. Distal arthrogryposis type 1 is characterized largely by camptodactyly and clubfoot. Hypoplasia and/or absence of some interphalangeal creases is common. The shoulders and hips are less frequently affected.
Variantes genéticas (ClinVar)
718 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 313 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Artrogripose distal tipo 1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
2 ensaios clínicos encontrados.
Publicações mais relevantes
Compound heterozygous variants in MYBPC1 lead to severe distal arthrogryposis type-1 manifestations.
Dominant missense variants in MYBPC1 encoding slow Myosin Binding Protein-C (sMyBP-C) have been increasingly linked to arthrogryposis syndromes and congenital myopathy with tremor. Herein, we describe novel compound heterozygous variants - NM_002465.4:[c.2486_2492del];[c.2663A > G] - present in fibronectin-III (Fn-III) C7 and immunoglobulin (Ig) C8 domains, respectively, manifesting as severe, early-onset distal arthrogryposis type-1, with the carrier requiring intensive care and several surgical interventions at an early age. Computational modeling predicts that the c.2486_2492del p.(Lys829IlefsTer7) variant destabilizes the structure of the Fn-III C7 domain, while the c.2663A > G p.(Asp888Gly) variant causes minimal structural alterations in the Ig C8 domain. Although the parents of the proband are heterozygous carriers for a single variant, they exhibit no musculoskeletal defects, suggesting a complex interplay between the two mutant alleles underlying this disorder. As emerging novel variants in MYBPC1 are shown to be causatively associated with musculoskeletal disease, it becomes clear that MYBPC1 should be included in relevant genetic screenings.
TNNI1 Mutated in Autosomal Dominant Proximal Arthrogryposis.
The main objective of this case report is to identify a gene associated with a Japanese family with autosomal dominant arthrogryposis. We performed clinicopathologic diagnosis and genomic analysis using trio-based exome sequencing. A 14-year-old boy had contractures in the proximal joints, and the serum creatine kinase level was elevated. Muscle biopsy demonstrated a moth-eaten appearance in some type 1 fibers, and electron microscopic analysis revealed that type 1 fibers had Z disk streaming. We identified a heterozygous nonsense variant, c.523A>T (p.K175*), in TNNI1 in the family. The altered amino acid residue is within the tropomyosin-binding site near the C-terminus, in a region homologous to the variational hotspot of Troponin I2 (TNNI2), which is associated with distal arthrogryposis type 1 and 2b. Compared with patients with TNNI2 variants, our patient had a milder phenotype and proximal arthrogryposis. We report here a case of proximal arthrogryposis associated with a TNNI1 nonsense variant, which expands the genetic and clinical spectrum of this disease. Further functional and genetic studies are required to clarify the role of TNNI1 in the disease.
Findings, Phenotypes, Diagnostic Accuracy, and Treatment in Freeman-Burian Syndrome.
Freeman-Burian syndrome (FBS) is a rare congenital myopathic craniofacial syndrome. Since publication of the genotype-correlated clinical diagnostic criteria, no complete survey of the literature has been accomplished. As part of the clinical practice guideline development, we evaluate diagnostic accuracy for FBS from 1938 to 2019 and range of findings, complications, treatments, and outcomes. Published manuscripts in PubMed, Google Scholar, and OMIM describing cases with a reported diagnosis of FBS, Sheldon-Hall syndrome, and distal arthrogryposes type 1 and 3 are initially included. Articles with sufficient case-level data for diagnosis verification are analyzed further. Of 724 unique papers considered, 188 papers describing 304 unique patients are included; 101 papers and 119 patients reflect an FBS diagnosis, with 80 patients meeting the full diagnostic criteria. Most cases are re-screened as distal arthrogryposis type 1. Among all cases re-screened as FBS, the presence of FBS pathognomonic craniofacial findings is not correlated with other physical findings. There are no significant differences between patients meeting the full diagnostic criteria and those not, but both are distinct from other diagnoses. Plastic surgery demonstrates the highest cumulative diagnostic accuracy for FBS overall (86.66%), while orthopedic surgery shows the lowest (44.83%). No statistically usable treatment-related or psychosocial data are available. Quality of case reports and patient data vary widely, reducing the statistical strength and significance. Major knowledge gaps exist in treatment, psychosocial, and longitudinal outcomes. At this point, it is impossible to derive clinical practice guidelines exclusively from the literature.
Findings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: protocol for systematic review and patient-level data meta-analysis.
Freeman-Sheldon and Sheldon-Hall syndromes (FSS and SHS) and distal arthrogryposis types 1 and 3 (DA1 and DA3) are rare, often confused, congenital syndromes. Few studies exist. With reported diagnosis unreliable, it would be scientifically inappropriate to consider articles describing FSS, SHS, DA1, or DA3, unless diagnoses were independently verified, rendering conventional systematic review and meta-analysis methodology inappropriate and necessitating patient-level data analysis (PROSPERO: CRD42015024740). As part of a clinical practise guideline development process, we evaluate (1) diagnostic accuracy from 1938-2017, using the Stevenson criteria; (2) the most common physical findings, possible frequency clusters, and complications of physical findings amongst patients with FSS; and (3) treatment types and outcomes. All papers reporting diagnosis of FSS, SHS, DA1, and DA3 are included in searching PubMed and Google Scholar from December 2014 to July 2015 and again before final analyses. Patients with FSS are divided into four phenotype-defined sub-types; all patients are grouped by published diagnosis and medical speciality. Significance of physical findings and historical data is evaluated by chi-square. Associations of physical findings and history with diagnosis and treatment outcome are evaluated by Pearson correlation and linear regression analysis. Two-tailed alpha level of 0.05 is used throughout. The need for detailed patient-level data extraction may limit the types of articles included and questions able to be answered. For treatment and psychosocial health outcomes, we anticipate enhanced difficulties, which may limit significance, power, and results' usability. We hope to outline knowledge gaps and prioritise areas for clinical investigation. CRD42015024740 Universal Trial Number: U1111-1172-4670.
A novel missense mutation of TNNI2 in a Chinese family cause distal arthrogryposis type 1.
The distal arthrogryposis (DA) syndromes are a group of disorders characterized by congenital contractures of limbs. According to phenotypical characteristics, DA syndromes have been clinically classified into 10 types. Currently, at least nine disease causing genes have been identified for different types of DA. Here, we report a 3-generation Chinese pedigree with three DA affected members. We performed whole exome sequencing on two affected and one unaffected individuals of this family and successfully identified a novel missense mutation in TNNI2 as the pathogenic mutation. The TNNI2 gene encodes a subunit of the troponin complex, a contractile machinery of the muscle. The mutation p.F178C that could change the H-bond formation of a neighboring residue occurs at a highly conserved position, suggesting that this variation probably affects the TNNI2 protein function. Our study also demonstrates the power of whole exome sequencing in causal mutation identification for phenotypically variable and genetically heterogeneous disorders.
Publicações recentes
Compound heterozygous variants in MYBPC1 lead to severe distal arthrogryposis type-1 manifestations.
TNNI1 Mutated in Autosomal Dominant Proximal Arthrogryposis.
Findings, Phenotypes, Diagnostic Accuracy, and Treatment in Freeman-Burian Syndrome.
Findings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: protocol for systematic review and patient-level data meta-analysis.
Freeman-Sheldon syndrome in a 29-year-old woman presenting with rare and previously undescribed features.
📚 EuropePMC151 artigos no totalmostrando 6
Compound heterozygous variants in MYBPC1 lead to severe distal arthrogryposis type-1 manifestations.
GeneTNNI1 Mutated in Autosomal Dominant Proximal Arthrogryposis.
Neurology. GeneticsFindings, Phenotypes, Diagnostic Accuracy, and Treatment in Freeman-Burian Syndrome.
The Journal of craniofacial surgeryFindings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: protocol for systematic review and patient-level data meta-analysis.
Systematic reviewsFreeman-Sheldon syndrome in a 29-year-old woman presenting with rare and previously undescribed features.
BMJ case reportsA novel missense mutation of TNNI2 in a Chinese family cause distal arthrogryposis type 1.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Artrogripose distal tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Artrogripose distal tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Compound heterozygous variants in MYBPC1 lead to severe distal arthrogryposis type-1 manifestations.
- TNNI1 Mutated in Autosomal Dominant Proximal Arthrogryposis.
- Findings, Phenotypes, Diagnostic Accuracy, and Treatment in Freeman-Burian Syndrome.
- Findings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: protocol for systematic review and patient-level data meta-analysis.
- A novel missense mutation of TNNI2 in a Chinese family cause distal arthrogryposis type 1.
- Freeman-Sheldon syndrome in a 29-year-old woman presenting with rare and previously undescribed features.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1146(Orphanet)
- MONDO:0015240(MONDO)
- GARD:787(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56013702(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
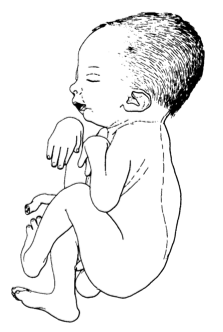
Artrogripose distal tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata