A artrogripose múltipla congênita neurogênica é uma forma de artrogripose múltipla congênita caracterizada por imobilidade congênita dos membros com fixação de múltiplas articulações e perda muscular. Esta condição é secundária à atrofia muscular neurogênica.
Introdução
O que você precisa saber de cara
A artrogripose múltipla congênita neurogênica é uma forma de artrogripose múltipla congênita caracterizada por imobilidade congênita dos membros com fixação de múltiplas articulações e perda muscular. Esta condição é secundária à atrofia muscular neurogênica.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 30 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 97 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisInhibits fibrillization of amyloid-beta peptide during the elongation phase. Has also been shown to assemble amyloid fibrils into protease-resistant aggregates. Binds heparin
Membrane
Fibrosis of extraocular muscles, congenital, 5
An ocular motility disorder characterized by congenital dysinnervation of various cranial nerves to ocular muscles. Clinical features are ophthalmoplegia, anchoring of the eyes in downward gaze, ptosis, and backward tilt of the head.
Proposed to be involved in endosomal maturation implicating in part VPS33B. In epithelial cells, the VPS33B:VIPAS39 complex may play a role in the apical RAB11A-dependent recycling pathway and in the maintenance of the apical-basolateral polarity (PubMed:20190753). May play a role in lysosomal trafficking, probably via association with the core HOPS complex in a discrete population of endosomes; the functions seems to be independent of VPS33B (PubMed:19109425). May play a role in vesicular traff
CytoplasmCytoplasmic vesicleEarly endosomeRecycling endosomeLate endosome
Arthrogryposis, renal dysfunction and cholestasis syndrome 2
A multisystem disorder, characterized by neurogenic arthrogryposis multiplex congenita, renal tubular dysfunction and neonatal cholestasis with bile duct hypoplasia and low gamma glutamyl transpeptidase activity. Platelet dysfunction is common.
May play a role in vesicle-mediated protein trafficking to lysosomal compartments and in membrane docking/fusion reactions of late endosomes/lysosomes. Required for proper trafficking and targeting of the collagen-modifying enzyme lysyl hydroxylase 3 (LH3) to intracellular collagen (PubMed:28017832). Mediates phagolysosomal fusion in macrophages (PubMed:18474358). Proposed to be involved in endosomal maturation implicating VIPAS39. In epithelial cells, the VPS33B:VIPAS39 complex may play a role
Late endosome membraneLysosome membraneEarly endosomeCytoplasmic vesicle, clathrin-coated vesicleRecycling endosome
Arthrogryposis, renal dysfunction, and cholestasis 1
An autosomal recessive multisystem disorder with characteristics of congenital joint contractures, renal tubular dysfunction, neonatal cholestasis with bile duct hypoplasia and low gamma glutamyl transpeptidase activity, severe failure to thrive, ichthyosis, and a defect in platelet alpha-granule biogenesis. Most patients do not survive past the first year of life.
Possible role in transport between endoplasmic reticulum and Golgi
Endoplasmic reticulum membraneEndoplasmic reticulum-Golgi intermediate compartment membraneGolgi apparatus membrane
Arthrogryposis multiplex congenita 2, neurogenic type
A form of arthrogryposis multiplex congenita, a heterogeneous group of disorders characterized by multiple joint contractures resulting, in some cases, from reduced or absent fetal movements. AMC2 is due to a neurogenic defect and is characterized by congenital immobility of the limbs with fixation of multiple joints, and muscle wasting. AMC2 transmission pattern is consistent with autosomal recessive inheritance in several families. Penetrance may be incomplete in females.
Component of the AP2-containing clathrin coat that may regulate clathrin-dependent trafficking at plasma membrane, TGN and endosomal system (Probable). A possible serine/threonine-protein kinase toward the beta2-subunit of the plasma membrane adapter complex AP2 and other proteins in presence of poly-L-lysine has not been confirmed (PubMed:15809293, PubMed:16914521). By regulating the expression of excitatory receptors at synapses, plays an essential role in neuronal function and signaling and i
Cytoplasmic vesicle, clathrin-coated vesicleGolgi apparatus, trans-Golgi network membraneEndosome membrane
Arthrogryposis multiplex congenita 4, neurogenic, with agenesis of the corpus callosum
A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC4 is an autosomal recessive, severe form with onset in utero. Patients manifest little or no fetal movements, significant contractures affecting the upper and lower limbs, dysmorphic facial features, optic atrophy, limb fractures, profound global developmental delay, seizures, and peripheral neuropathy. Many patients die in early childhood.
Variantes genéticas (ClinVar)
212 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Artrogripose múltipla congênita tipo neurogênico
Centros de Referência SUS
24 centros habilitados pelo SUS para Artrogripose múltipla congênita tipo neurogênico
Centros para Artrogripose múltipla congênita tipo neurogênico
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A Case Report: Can Prioritizing Sensory Integration Therapy Help Improve Gross Motor Function in a Rare Case of Neurogenic Arthrogryposis Multiplex Congenita?
Arthrogryposis multiplex congenital (AMC) is a congenital disorder diagnosed with extremity contractures, restricted joint range of motion, foot abnormalities, and hip dislocation. The current literature emphasizes medical and surgical management, but very few studies provide insight into physiotherapy management for AMC. We reported the case of a 16-month-old male diagnosed with AMC, operated on both hips for teratologic dislocation. Physiotherapy examination was conducted, and treatment was planned based on the principles of Sensory Integration Therapy (SIT) and neurodevelopmental technique (NDT) with orthosis to assist in functional recovery. He achieved motor milestones within one year of regular physiotherapy treatment. As per our literature search, this is the first study where an attempt has been made to utilize sensory integration along with NDT for the treatment of AMC, although the clinical presentation of the patient shows more musculoskeletal abnormalities.
DST variants are responsible for neurogenic arthrogryposis multiplex congenita enlarging the spectrum of type VI hereditary sensory autonomic neuropathy.
Arthrogryposis multiplex congenita (AMC) is a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. Through whole-exome sequencing combined with arrayCGH from DNA of a fetus presenting with early onset AMC, we identified biallelic loss of function variants in Dystonin (DST): a stop gain variant (NM_001144769.5:c.12208G > T:p.(Glu4070Ter)) on the neuronal isoform and a 175 kb microdeletion including exons 25-96 of this isoform on the other allele [NC_000006.11:g.(56212278_56323554)_(56499398_56507586)del]. Transmission electron microscopy of the sciatic nerve revealed abnormal morphology of the peripheral nerve with severe hypomyelination associated with dramatic reduction of fiber density which highlights the critical role of DST in peripheral nerve axonogenesis during development in human. Variants in the neuronal isoforms of DST cause hereditary sensory and autonomic neuropathy which has been reported in several unrelated families with highly variable age of onset from fetal to adult onset. Our data enlarge the disease mechanisms of neurogenic AMC.
Early onset hereditary neuronopathies: an update on non-5q motor neuron diseases.
Hereditary motor neuropathies (HMN) were first defined as a group of neuromuscular disorders characterized by lower motor neuron dysfunction, slowly progressive length-dependent distal muscle weakness and atrophy, without sensory involvement. Their cumulative estimated prevalence is 2.14/100 000 and, to date, around 30 causative genes have been identified with autosomal dominant, recessive,and X-linked inheritance. Despite the advances of next generation sequencing, more than 60% of patients with HMN remain genetically uncharacterized. Of note, we are increasingly aware of the broad range of phenotypes caused by pathogenic variants in the same gene and of the considerable clinical and genetic overlap between HMN and other conditions, such as Charcot-Marie-Tooth type 2 (axonal), spinal muscular atrophy with lower extremities predominance, neurogenic arthrogryposis multiplex congenita and juvenile amyotrophic lateral sclerosis. Considering that most HMN present during childhood, in this review we primarily aim to summarize key clinical features of paediatric forms, including recent data on novel phenotypes, to help guide differential diagnosis and genetic testing. Second, we describe newly identified causative genes and molecular mechanisms, and discuss how the discovery of these is changing the paradigm through which we approach this group of conditions.
A reverse genetics and genomics approach to gene paralog function and disease: Myokymia and the juxtaparanode.
The leucine-rich glioma-inactivated (LGI) family consists of four highly conserved paralogous genes, LGI1-4, that are highly expressed in mammalian central and/or peripheral nervous systems. LGI1 antibodies are detected in subjects with autoimmune limbic encephalitis and peripheral nerve hyperexcitability syndromes (PNHSs) such as Isaacs and Morvan syndromes. Pathogenic variations of LGI1 and LGI4 are associated with neurological disorders as disease traits including familial temporal lobe epilepsy and neurogenic arthrogryposis multiplex congenita 1 with myelin defects, respectively. No human disease has been reported associated with either LGI2 or LGI3. We implemented exome sequencing and family-based genomics to identify individuals with deleterious variants in LGI3 and utilized GeneMatcher to connect practitioners and researchers worldwide to investigate the clinical and electrophysiological phenotype in affected subjects. We also generated Lgi3-null mice and performed peripheral nerve dissection and immunohistochemistry to examine the juxtaparanode LGI3 microarchitecture. As a result, we identified 16 individuals from eight unrelated families with loss-of-function (LoF) bi-allelic variants in LGI3. Deep phenotypic characterization showed LGI3 LoF causes a potentially clinically recognizable PNHS trait characterized by global developmental delay, intellectual disability, distal deformities with diminished reflexes, visible facial myokymia, and distinctive electromyographic features suggestive of motor nerve instability. Lgi3-null mice showed reduced and mis-localized Kv1 channel complexes in myelinated peripheral axons. Our data demonstrate bi-allelic LoF variants in LGI3 cause a clinically distinguishable disease trait of PNHS, most likely caused by disturbed Kv1 channel distribution in the absence of LGI3.
Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita.
Pathogenic variants in the X-linked gene ZC4H2, which encodes a zinc-finger protein, cause an infrequently described syndromic form of arthrogryposis multiplex congenita (AMC) with central and peripheral nervous system involvement. We present genetic and detailed phenotypic information on 23 newly identified families and simplex cases that include 19 affected females from 18 families and 14 affected males from nine families. Of note, the 15 females with deleterious de novo ZC4H2 variants presented with phenotypes ranging from mild to severe, and their clinical features overlapped with those seen in affected males. By contrast, of the nine carrier females with inherited ZC4H2 missense variants that were deleterious in affected male relatives, four were symptomatic. We also compared clinical phenotypes with previously published cases of both sexes and provide an overview on 48 males and 57 females from 42 families. The spectrum of ZC4H2 defects comprises novel and recurrent mostly inherited missense variants in affected males, and de novo splicing, frameshift, nonsense, and partial ZC4H2 deletions in affected females. Pathogenicity of two newly identified missense variants was further supported by studies in zebrafish. We propose ZC4H2 as a good candidate for early genetic testing of males and females with a clinical suspicion of fetal hypo-/akinesia and/or (neurogenic) AMC.
Publicações recentes
A Case Report: Can Prioritizing Sensory Integration Therapy Help Improve Gross Motor Function in a Rare Case of Neurogenic Arthrogryposis Multiplex Congenita?
DST variants are responsible for neurogenic arthrogryposis multiplex congenita enlarging the spectrum of type VI hereditary sensory autonomic neuropathy.
Early onset hereditary neuronopathies: an update on non-5q motor neuron diseases.
A reverse genetics and genomics approach to gene paralog function and disease: Myokymia and the juxtaparanode.
Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita.
📚 EuropePMC6 artigos no totalmostrando 5
A Case Report: Can Prioritizing Sensory Integration Therapy Help Improve Gross Motor Function in a Rare Case of Neurogenic Arthrogryposis Multiplex Congenita?
CureusDST variants are responsible for neurogenic arthrogryposis multiplex congenita enlarging the spectrum of type VI hereditary sensory autonomic neuropathy.
Clinical geneticsEarly onset hereditary neuronopathies: an update on non-5q motor neuron diseases.
Brain : a journal of neurologyA reverse genetics and genomics approach to gene paralog function and disease: Myokymia and the juxtaparanode.
American journal of human geneticsDeleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita.
Human mutationAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Artrogripose múltipla congênita tipo neurogênico.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Artrogripose múltipla congênita tipo neurogênico
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A Case Report: Can Prioritizing Sensory Integration Therapy Help Improve Gross Motor Function in a Rare Case of Neurogenic Arthrogryposis Multiplex Congenita?
- DST variants are responsible for neurogenic arthrogryposis multiplex congenita enlarging the spectrum of type VI hereditary sensory autonomic neuropathy.
- Early onset hereditary neuronopathies: an update on non-5q motor neuron diseases.
- A reverse genetics and genomics approach to gene paralog function and disease: Myokymia and the juxtaparanode.
- Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1143(Orphanet)
- OMIM OMIM:208100(OMIM)
- MONDO:0008823(MONDO)
- GARD:790(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q708165(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
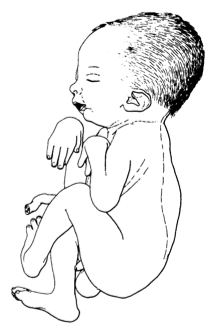
Artrogripose múltipla congênita tipo neurogênico
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata