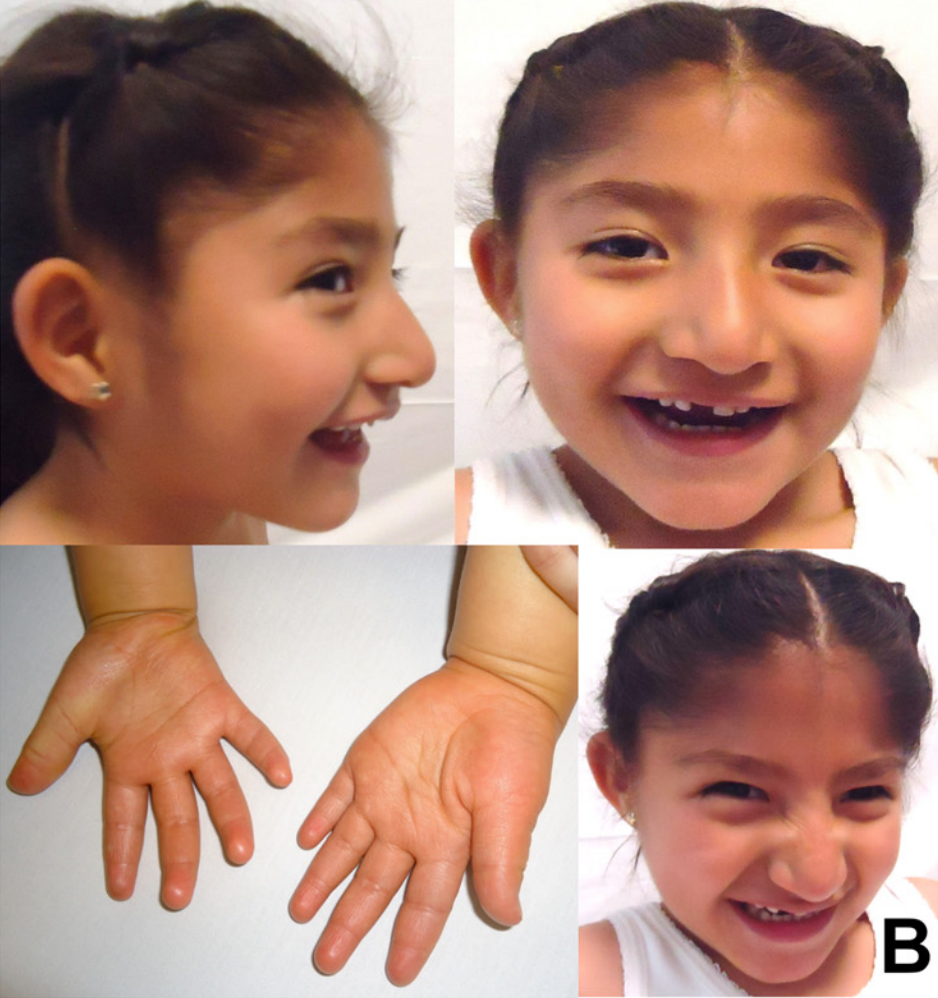
Síndrome de Angelman por dissomia uniparental paterna do cromossomo 15, associada a atraso global do desenvolvimento, deficiência intelectual e características faciais como prognatismo mandibular. Pode apresentar dificuldades alimentares, hiperreflexia e crises epilépticas.
Introdução
O que você precisa saber de cara
Visão geral
A Dissomia uniparental de origem paterna, cromossomo 15 é uma condição genética rara que leva à síndrome de Angelman. Ela ocorre quando uma pessoa herda duas cópias do cromossomo 15 do pai e nenhuma da mãe. Isso afeta a expressão do gene UBE3A, essencial para o desenvolvimento neurológico normal. A condição se manifesta na primeira infância e pode causar atrasos no desenvolvimento, convulsões e outras alterações neurológicas.[1][3]
Sinais e sintomas
Os sinais e sintomas da Dissomia uniparental de origem paterna, cromossomo 15 incluem dificuldades alimentares, sucção pobre, hipotonia do lactente (tônus muscular baixo), atraso global do desenvolvimento, microcefalia secundária (cessação do crescimento da cabeça), deficiência intelectual, convulsões (como crise tônico-clônica bilateral, crise de ausência atípica), anormalidades no EEG, fala pobre ou ausente, protrusão da língua, prognatismo mandibular (queixo proeminente), dentes amplamente espaçados, marcha de base alargada, hiperreflexia dos membros inferiores, hipopigmentação da pele e do cabelo, disfagia (dificuldade para engolir) e intolerância ao calor.[1][3]
Causas genéticas
A condição é causada pela dissomia uniparental paterna do cromossomo 15, ou seja, a pessoa herda duas cópias desse cromossomo do pai e nenhuma da mãe. Isso resulta na perda da expressão do gene UBE3A (Ubiquitin-protein ligase E3A), localizado nesse cromossomo. Esse gene é responsável por produzir uma proteína importante para a função cerebral normal. A ausência da cópia materna ativa do gene UBE3A leva aos sintomas observados.[1][4]
Diagnóstico
O diagnóstico da Dissomia uniparental de origem paterna, cromossomo 15 é realizado por meio de testes genéticos. Os procedimentos disponíveis no SUS incluem cariótipo com bandas G, Q ou R, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES) e dosagem de alfa-fetoproteína. Além disso, há 761 variantes registradas no ClinVar relacionadas a essa condição, e o atendimento em reabilitação para doenças raras também está previsto.[1][4]
Tratamento e manejo
O manejo da Dissomia uniparental de origem paterna, cromossomo 15 é multidisciplinar e visa controlar os sintomas e melhorar a qualidade de vida. Inclui acompanhamento neurológico para convulsões, suporte nutricional para dificuldades alimentares e disfagia, fisioterapia para hipotonia e alterações de marcha, fonoaudiologia para problemas de fala e deglutição, e terapia ocupacional. O atendimento em reabilitação para doenças raras está disponível no SUS. Não há medicamentos específicos listados para esta condição.[1]
Prognóstico e qualidade de vida
O prognóstico da Dissomia uniparental de origem paterna, cromossomo 15 varia conforme a gravidade dos sintomas. A deficiência intelectual e as convulsões podem persistir ao longo da vida, exigindo cuidados contínuos. Com intervenções precoces e suporte multidisciplinar, é possível melhorar a qualidade de vida, mas a condição geralmente requer acompanhamento médico e terapêutico de longo prazo.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome de Angelman por dissomia uniparental paterna do cromossomo 15, associada a atraso global do desenvolvimento, deficiência intelectual e características faciais como prognatismo mandibular. Pode apresentar dificuldades alimentares, hiperreflexia e crises epilépticas.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Dissomia uniparental de origem paterna, cromossomo 15 é uma condição genética rara que leva à síndrome de Angelman. Ela ocorre quando uma pessoa herda duas cópias do cromossomo 15 do pai e nenhuma da mãe. Isso afeta a expressão do gene UBE3A, essencial para o desenvolvimento neurológico normal. A condição se manifesta na primeira infância e pode causar atrasos no desenvolvimento, convulsões e outras alterações neurológicas.[1][3]
Sinais e sintomas
Os sinais e sintomas da Dissomia uniparental de origem paterna, cromossomo 15 incluem dificuldades alimentares, sucção pobre, hipotonia do lactente (tônus muscular baixo), atraso global do desenvolvimento, microcefalia secundária (cessação do crescimento da cabeça), deficiência intelectual, convulsões (como crise tônico-clônica bilateral, crise de ausência atípica), anormalidades no EEG, fala pobre ou ausente, protrusão da língua, prognatismo mandibular (queixo proeminente), dentes amplamente espaçados, marcha de base alargada, hiperreflexia dos membros inferiores, hipopigmentação da pele e do cabelo, disfagia (dificuldade para engolir) e intolerância ao calor.[1][3]
Causas genéticas
A condição é causada pela dissomia uniparental paterna do cromossomo 15, ou seja, a pessoa herda duas cópias desse cromossomo do pai e nenhuma da mãe. Isso resulta na perda da expressão do gene UBE3A (Ubiquitin-protein ligase E3A), localizado nesse cromossomo. Esse gene é responsável por produzir uma proteína importante para a função cerebral normal. A ausência da cópia materna ativa do gene UBE3A leva aos sintomas observados.[1][4]
Diagnóstico
O diagnóstico da Dissomia uniparental de origem paterna, cromossomo 15 é realizado por meio de testes genéticos. Os procedimentos disponíveis no SUS incluem cariótipo com bandas G, Q ou R, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES) e dosagem de alfa-fetoproteína. Além disso, há 761 variantes registradas no ClinVar relacionadas a essa condição, e o atendimento em reabilitação para doenças raras também está previsto.[1][4]
Tratamento e manejo
O manejo da Dissomia uniparental de origem paterna, cromossomo 15 é multidisciplinar e visa controlar os sintomas e melhorar a qualidade de vida. Inclui acompanhamento neurológico para convulsões, suporte nutricional para dificuldades alimentares e disfagia, fisioterapia para hipotonia e alterações de marcha, fonoaudiologia para problemas de fala e deglutição, e terapia ocupacional. O atendimento em reabilitação para doenças raras está disponível no SUS. Não há medicamentos específicos listados para esta condição.[1]
Prognóstico e qualidade de vida
O prognóstico da Dissomia uniparental de origem paterna, cromossomo 15 varia conforme a gravidade dos sintomas. A deficiência intelectual e as convulsões podem persistir ao longo da vida, exigindo cuidados contínuos. Com intervenções precoces e suporte multidisciplinar, é possível melhorar a qualidade de vida, mas a condição geralmente requer acompanhamento médico e terapêutico de longo prazo.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 28 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
E3 ubiquitin-protein ligase which accepts ubiquitin from an E2 ubiquitin-conjugating enzyme in the form of a thioester and transfers it to its substrates (PubMed:10373495, PubMed:16772533, PubMed:19204938, PubMed:19233847, PubMed:19325566, PubMed:19591933, PubMed:22645313, PubMed:24273172, PubMed:24728990, PubMed:30020076). Several substrates have been identified including the BMAL1, ARC, LAMTOR1, RAD23A and RAD23B, MCM7 (which is involved in DNA replication), annexin A1, the PML tumor suppresso
CytoplasmNucleus
Angelman syndrome
A neurodevelopmental disorder characterized by severe motor and intellectual retardation, ataxia, frequent jerky limb movements and flapping of the arms and hands, hypotonia, seizures, absence of speech, frequent smiling and episodes of paroxysmal laughter, open-mouthed expression revealing the tongue.
Variantes genéticas (ClinVar)
761 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Dissomia uniparental de origem paterna, cromossomo 15
Centros de Referência SUS
24 centros habilitados pelo SUS para Dissomia uniparental de origem paterna, cromossomo 15
Centros para Dissomia uniparental de origem paterna, cromossomo 15
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A stem cell-based toolkit to model Angelman syndrome caused by paternal uniparental disomy of chromosome 15.
Angelman syndrome is a rare neurodevelopmental disorder caused by the loss of function of the maternally inherited UBE3A gene within the chr15q11-q13 region. This gene is subjected to a tissue-specific form of genomic imprinting leading to the silencing of the paternal allele in neurons. Angelman syndrome can result from various (epi)genetic mechanisms, with paternal uniparental disomy of chromosome 15 (patUPD15) being one of the rarest and least studied due to the absence of suitable models. To address this gap, we generated three independent induced pluripotent stem cell (iPSC) lines from individuals with Angelman syndrome caused by patUPD15, alongside genetically matched unaffected familial controls. Peripheral blood mononuclear cells (PBMCs) were reprogrammed into iPSCs using a non-integrative Sendai virus-based approach expressing the Yamanaka factors. All iPSC lines underwent rigorous quality control, confirming stem cell identity, trilineage differentiation potential, and genetic and epigenetic integrity. This newly established iPSC toolkit provides a powerful platform to investigate the molecular underpinnings of Angelman syndrome caused by patUPD15, paving the way for future translational research and therapeutic development tailored for this understudied form of the disorder.
UBE3A reinstatement restores behaviorand proteome in an Angelman syndrome mouse model of imprinting defects.
Angelman Syndrome (AS) is a severe neurodevelopmental disorder with only symptomatic treatment currently available. The primary cause of AS is loss of functional UBE3A protein. This can be caused by deletions in the maternal 15q11-q13 region, maternal AS-imprinting center defects (mICD), paternal uniparental disomy of chromosome 15 (UPD) or mutations within the UBE3A gene. Current mouse models are Ube3a-centric and do not address expression changes of other genes in the 15q11-q13 locus on the pathophysiology of AS. This limits the ability to discern differences in therapeutic responses to current UBE3A-targeting strategies and hampers the identification of novel therapeutics/co-therapeutics. Using a mouse line that harbors a maternally inherited mutation affecting the AS-PWS imprinting center ('mICD mice'), we studied the impact of the mICD or UPD AS subtype on behavior, seizure susceptibility and proteome. Additionally, by using mice overexpressing two copies of Ube3a or antisense oligonucleotide (ASO) targeting Ube3a-ATS, we analyzed the impact of bi-allelic Ube3a activation on behavior and proteome. mICD mice showed 80% reduction in UBE3A protein, bi-allelic expression of Ube3a-ATS and Mkrn3-Snord115 gene cluster, leading to robust AS behavioral deficits and proteome alterations similar to Ube3am-/p+ mice. Genetic UBE3A overexpression in mICD mice, mimicking therapeutic strategies that effectively activate the biallelic silenced Ube3a gene, resulted in a complete rescue of all behavioral phenotypes, seizure susceptibility and proteome alterations. Subsequently, treatment with an antisense oligonucleotide (ASO) to directly activate the biallelic silenced Ube3a gene in mICD mice also resulted in efficient reinstatement of UBE3A, 30% higher relative to WT, alongside a partial rescue of behavioral phenotypes. Despite using a highly robust AS-specific behavioral battery, we did not investigate readouts such as neuronal activity and sleep, for which impairments in Ube3am-/p+ mice were described. Taken together, these findings demonstrate that the loss of UBE3A protein is the primary factor underlying AS phenotypes in this mICD/UPD mouse model of AS, while the biallelic expressed genes in this locus play either a marginal or yet unidentified role. These findings also corroborate UBE3A reinstatement as an attractive therapeutic strategy for AS individuals carrying an mICD or UPD mutation.
Paternal UPD (15) With Disease-Causing Mutation and Small Supernumerary Ring Chromosome 15: A Case Report.
Uniparental disomy (UPD) constitutes an unconventional mode of inheritance that disrupts the typical biparental genetic contribution and may result in phenotypic abnormalities. This report centers on a patient diagnosed with Bartter syndrome Type 1, attributed to a homozygous pathogenic variant in SLC12A1 unmasked by mosaic paternal UPD of chromosome 15. We hypothesize that this pattern (or constellation) emerged from a trisomy rescue event, resulting in two distinct cell lines. Concurrently, the unmasking of a pathogenic paternal SLC12A1 variant by trisomy rescue resulted in the manifestation of Bartter syndrome Type 1. The maternally derived ring chromosome 15 and its impact on nondisjunction and UPD elucidate a unique etiology of Bartter syndrome. Furthermore, the presence of a pathogenic paternal SLC12A1 variant underscores the pivotal role of trisomic rescue and paternal UPD in unveiling a recessive variant.
Bone health in children with Angelman syndrome at the ENCORE Expertise Center.
Angelman syndrome (AS) is a rare genetic disorder due to lack of UBE3A function on chromosome 15q11.2q13 caused by a deletion, uniparental paternal disomy (UPD), imprinting center disorder (ICD), or pathological variant of the UBE3A gene. AS is characterized by developmental delay, epilepsy, and lack of speech. Although fractures are observed frequently in our clinical practice, there are few studies on bone health in AS. The aim of this study is to investigate bone health in children with AS. In this prospective cohort study, we describe bone health in 91 children with AS visiting the ENCORE Expertise Center for AS between April 2010 and December 2021. Bone health was assessed with the bone health index (BHI) in standard deviation score (SDS) measured by digital radiogrammetry of the left hand using BoneXpert software. Risk factors analyzed were age, sex, genetic subtype, epilepsy, anti-seizure medication use, mobility, body mass index (BMI), and onset of puberty. Children with AS had a mean BHI of -1.77 SDS (SD 1.4). A significantly lower BHI was found in children with a deletion (-2.24 SDS) versus non-deletion (-1.02 SDS). Other factors associated with reduced BHI-SDS were inability to walk and late onset of puberty. Children with a history of one or more fractures (22%) had a significantly lower BHI than children without fractures (-2.60 vs -1.56 SDS). Longitudinal analysis showed a significant decrease in BHI-SDS with age in all genetic subtypes. Conclusions: Children with AS have a reduced bone health. Risk factors are deletion genotype, no independent walking, and late onset of puberty. Bone health decreased significantly with age. What is Known: • Children with neurological disorders often have a low bone health and higher risk of fractures. • Little is known about bone health in children with Angelman syndrome (AS). What is New: • Children with AS showed a reduced bone health and this was significantly associated with having a deletion, not being able to walk independently, and late onset of puberty. • Longitudinal analysis showed a significant decrease in bone health as children got older.
Relationship of thyroid function with genetic subtypes and treatment with growth hormone in Prader-Willi syndrome.
Prader-Willi syndrome (PWS) is the most common genetic syndrome with obesity and results from loss of expression of paternally inherited genes on chromosome 15q11-q13 by a variety of mechanisms which include large deletions (70%-75%), maternal uniparental disomy (UPD) (20%-30%), and imprinting defects (2%-5%) or balanced translocations. Individuals often have a characteristic behavior disorder with mild intellectual disability, infantile hypotonia associated with poor sucking, short stature, and obesity. PWS is characterized by hypothalamic-pituitary axis dysfunction with growth hormone (GH) deficiency, hypogonadism, and several other hormonal deficiencies resulting in short stature, centrally driven excessive appetite (hyperphagia), central obesity, cryptorchidism, and decreased lean body mass. In this study, we determined and sought differences in the incidence of thyroid abnormalities among the common genetic subtypes in a cohort of 52 subjects with PWS because there was limited literature available. We also sought the effects of growth hormone (GH) treatment on the thyroid profile. Fifty-two subjects with a genetically confirmed diagnosis of PWS were included in this study at the University of California, Irvine. Blood samples for baseline thyroxine stimulating hormone (TSH) and free thyroxine (fT4) levels were obtained in the morning after an overnight fast for 8-12 h. Statistical analyses were performed with SPSS (SPSS Inc., 21.0). Mean values were analyzed by one-way ANOVA, and student's t-test and statistical significance were set at p < 0.05. The subjects included 26 males and 26 females with an age range of 3-38 years. There were 29 subjects with chromosome 15q11-q13 deletions and 23 with UPD; 28 were GH treated currently or in the past, and 24 never received GH. There was no significant difference in age or body mass index (BMI) (kg/m2) between GH-treated versus non-GH-treated groups. BMI was higher in the deletion group compared to the UPD group (p = 0.05). We identified two individuals who were clinically diagnosed and treated for hypothyroidism, one of whom was on GH supplements. We identified two additional individuals with subclinical hypothyroidism who were not on GH treatment, giving a frequency of 7.6% (4/52) in this cohort of patients. We did not find significant differences in thyroid function (TSH) in the deletion versus UPD groups. We found significant differences in thyroid function, however, between GH-treated and non-GH-treated groups. The mean TSH was lower (2.25 ± 1.17 uIU/M, range 0.03-4.92 uIU/M versus 2.80 ± 1.44 uIU/M, range 0.55-5.33 uIU/M respectively, p = 0.046), and the free T4 levels were significantly higher (1.13 ± 0.70 and 1.03 ± 0.11 ng/dL, respectively, p = 0.05) in the GH-treated individuals compared to non-GH-treated individuals. In this cohort of subjects with PWS, we identified two previously diagnosed individuals with hypothyroidism and two individuals with subclinical hypothyroidism (4/52, 7.6%), three of whom were not receiving GH treatment. We did not find any significant differences in thyroid function between molecular subtypes; however, we found that euthyroid status (lower TSH levels and higher free T4 levels) was significantly higher in individuals who were treated with GH compared to the untreated group. We recommend that individuals with PWS should be screened regularly for thyroid deficiency and start treatment early with GH in view of the potentially lower incidence of thyroid deficiency.
Publicações recentes
A stem cell-based toolkit to model Angelman syndrome caused by paternal uniparental disomy of chromosome 15.
Relationship of thyroid function with genetic subtypes and treatment with growth hormone in Prader-Willi syndrome.
Bone health in children with Angelman syndrome at the ENCORE Expertise Center.
Diagnosis of Prader-Willi syndrome and Angelman syndrome by targeted nanopore long-read sequencing.
Methylation analysis and developmental profile of two individuals with Angelman syndrome due to mosaic imprinting defects.
📚 EuropePMC1 artigos no totalmostrando 32
A stem cell-based toolkit to model Angelman syndrome caused by paternal uniparental disomy of chromosome 15.
Human cellUBE3A reinstatement restores behaviorand proteome in an Angelman syndrome mouse model of imprinting defects.
Molecular autismPaternal UPD (15) With Disease-Causing Mutation and Small Supernumerary Ring Chromosome 15: A Case Report.
Case reports in geneticsRelationship of thyroid function with genetic subtypes and treatment with growth hormone in Prader-Willi syndrome.
American journal of medical genetics. Part AGenetic investigation of the ubiquitin-protein ligase E3A gene as putative target in Angelman syndrome.
World journal of clinical casesBone health in children with Angelman syndrome at the ENCORE Expertise Center.
European journal of pediatricsPrader-Willi and Angelman Syndromes: Mechanisms and Management.
The application of clinical geneticsDiagnosis of Prader-Willi syndrome and Angelman syndrome by targeted nanopore long-read sequencing.
European journal of medical geneticsAngelman syndrome with mosaic paternal uniparental disomy suggestive of mitotic nondisjunction.
Journal of human geneticsMethylation analysis and developmental profile of two individuals with Angelman syndrome due to mosaic imprinting defects.
European journal of medical geneticsGenotype-Phenotype Correlations in Angelman Syndrome.
GenesBlended phenotype of combination of HERC2 and AP3B2 deficiency and Angelman syndrome caused by paternal isodisomy of chromosome 15.
American journal of medical genetics. Part APrader-Willi syndrome: reflections on seminal studies and future therapies.
Open biologyCongenital ichthyosis in Prader-Willi syndrome associated with maternal chromosome 15 uniparental disomy: Case report and review of autosomal recessive conditions unmasked by UPD.
American journal of medical genetics. Part AAngelman syndrome genotypes manifest varying degrees of clinical severity and developmental impairment.
Molecular psychiatryBlended phenotype of AP4E1 deficiency and Angelman syndrome caused by paternal isodisomy of chromosome 15.
Brain & developmentMosaic paternal uniparental isodisomy of 15q11-q13 region causing Angelman phenotype.
Clinical dysmorphologyWhole exome sequencing and methylation‑specific multiplex ligation‑dependent probe amplification applied to identify Angelman syndrome due to paternal uniparental disomy in two unrelated patients.
Molecular medicine reportsA rapid and accurate methylation-sensitive high-resolution melting analysis assay for the diagnosis of Prader Willi and Angelman patients.
Molecular genetics & genomic medicineGenetic testing for Prader-Willi syndrome and Angelman syndrome in the clinical practice of Guangdong Province, China.
Molecular cytogeneticsAngelman Syndrome-Affected Individual with a Numerically Normal Karyotype and Isodisomic Paternal Uniparental Disomy of Chromosome 15 due to Maternal Robertsonian Translocation (14;15) by Monosomy Rescue.
Cytogenetic and genome researchNovel intragenic deletions within the UBE3A gene in two unrelated patients with Angelman syndrome: case report and review of the literature.
BMC medical geneticsAngelman Syndrome due to a Maternally Inherited Intragenic Deletion Encompassing Exons 7 and 8 of the UBE3A Gene.
Cytogenetic and genome researchUniparental Disomy of Chromosome 15 in Two Cases by Chromosome Microarray: A Lesson Worth Thinking.
Cytogenetic and genome researchAngelman Syndrome Caused by Chromosomal Rearrangements: A Case Report of 46,XX,+der(13)t(13;15)(q14.1;q12)mat,-15 with an Atypical Phenotype and Review of the Literature.
Cytogenetic and genome researchAngelman syndrome - insights into a rare neurogenetic disorder.
Nature reviews. NeurologyDNA Methylation Profiling of Uniparental Disomy Subjects Provides a Map of Parental Epigenetic Bias in the Human Genome.
American journal of human geneticsApplicability of genetic polymorphism analysis for the diagnosis of Angelman syndrome and the correlation between language difficulties and disease phenotype.
Genetics and molecular research : GMRAngelman syndrome and isovaleric acidemia: What is the link?
Molecular genetics and metabolism reportsClinical Application of an Innovative Multiplex-Fluorescent-Labeled STRs Assay for Prader-Willi Syndrome and Angelman Syndrome.
PloS oneComparative molecular approaches in Prader-Willi syndrome diagnosis.
GenePrader-Willi syndrome: a review of clinical, genetic, and endocrine findings.
Journal of endocrinological investigationAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Dissomia uniparental de origem paterna, cromossomo 15.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Dissomia uniparental de origem paterna, cromossomo 15
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A stem cell-based toolkit to model Angelman syndrome caused by paternal uniparental disomy of chromosome 15.
- UBE3A reinstatement restores behaviorand proteome in an Angelman syndrome mouse model of imprinting defects.
- Paternal UPD (15) With Disease-Causing Mutation and Small Supernumerary Ring Chromosome 15: A Case Report.
- Bone health in children with Angelman syndrome at the ENCORE Expertise Center.
- Relationship of thyroid function with genetic subtypes and treatment with growth hormone in Prader-Willi syndrome.
- Diagnosis of Prader-Willi syndrome and Angelman syndrome by targeted nanopore long-read sequencing.
- Methylation analysis and developmental profile of two individuals with Angelman syndrome due to mosaic imprinting defects.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:98795(Orphanet)
- MONDO:0020303(MONDO)
- GARD:19578(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55789292(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Dissomia uniparental de origem paterna, cromossomo 15
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata