A deficiência da fosforilase do fígado, também conhecida como doença de depósito de glicogênio tipo 6b (ou doença de Hers, GSD 6b), é uma forma rara e benigna de doença de depósito de glicogênio.
Introdução
O que você precisa saber de cara
A deficiência da fosforilase do fígado, também conhecida como doença de depósito de glicogênio tipo 6b (ou doença de Hers, GSD 6b), é uma forma rara e benigna de doença de depósito de glicogênio.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 34 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisAllosteric enzyme that catalyzes the rate-limiting step in glycogen catabolism, the phosphorolytic cleavage of glycogen to produce glucose-1-phosphate, and plays a central role in maintaining cellular and organismal glucose homeostasis
Cytoplasm, cytosol
Glycogen storage disease 6
A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected.
Variantes genéticas (ClinVar)
124 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença de armazenamento de glicogênio por deficiência de glicogenofosforilase hepática
Centros de Referência SUS
21 centros habilitados pelo SUS para Doença de armazenamento de glicogênio por deficiência de glicogenofosforilase hepática
Centros para Doença de armazenamento de glicogênio por deficiência de glicogenofosforilase hepática
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
NUPAD / Faculdade de Medicina UFMG
Av. Prof. Alfredo Balena, 189 - 5 andar - Centro, Belo Horizonte - MG, 30130-100 · CNES 2183226
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital de Clínicas da Universidade Federal de Pernambuco
Av. Prof. Moraes Rego, 1235 - Cidade Universitária, Recife - PE, 50670-901 · CNES 2561492
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital Universitário Onofre Lopes (HUOL)
Av. Nilo Peçanha, 620 - Petrópolis, Natal - RN, 59012-300 · CNES 2408570
Atenção Especializada
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Instituto da Criança e do Adolescente (ICr-HCFMUSP)
Av. Dr. Enéas Carvalho de Aguiar, 647 - Cerqueira César, São Paulo - SP, 05403-000 · CNES 2081695
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Sodium taurocholate cotransporter polypeptide deficiency combined with novel PYGL mutations in glycogen storage disease type VI: a rare case report.
Both Sodium Taurocholate Cotransporting Polypeptide Deficiency (NTCPD) and Glycogen Storage Disease Type VI (GSD-VI) are autosomal recessive (AR) genetic disorders that affect liver metabolism in newborns. NTCPD is primarily caused by mutations in the Solute Carrier Family 10 Member 1 (SLC10A1) gene, resulting in decreased bile acid transport function, while GSD-VI is caused by mutations in the phosphorylase glycogen liver (PYGL) gene, leading to a deficiency of liver glycogen phosphorylase. Both diseases are rare, and there have been no previous reports of their coexistence in a single patient. This case report details the rare occurrence of NTCP and GSD-VI in a 2-year-11-month-old female child. The patient presented with mild jaundice, hepatomegaly, and growth retardation. After 1 year and 6 months of treatment and follow-up, the patient's liver function and growth showed significant improvement. This case highlights the importance of comprehensive genetic analysis in diagnosing complex metabolic disorders.
Glycogen storage disease type IX: Long-term follow-up of 52 patients from three European countries.
Glycogen storage disease type IX (GSD IX) arises from hepatic phosphorylase b kinase (PhK) deficiency attributable to pathogenic variants in the PHKA2, PHKB, and PHKG2 genes. This multicenter retrospective study evaluated clinical and biochemical data from 52 patients diagnosed across three European countries, with a median follow-up of 9.3 years (range: 1-49). In the cohort, 86.5% were classified as GSD IXa, whereas GSD IXb and IXc accounted for 7.7% and 3.8%, respectively; one diagnosis was based solely on enzymatic testing. Null variants in PHKA2 consistently resulted in severe PhK deficiency, whereas missense variants and in-frame deletions were associated with variable enzymatic impairment (8/19 tested cases). The median age at symptom onset was 1.6 years, and the mean age at diagnosis was 2.0 years. Predominant manifestations included hepatomegaly (82%), elevated aminotransferases (81%), hypertriglyceridemia (71%), hypercholesterolemia (67%), hypoglycemia (46%), hyperlactatemia (38%), and short stature (30%). Aberrant apolipoprotein C-III glycosylation was detected in 80% of analyzed samples. Nutritional intervention was associated with improved growth (height SD score - 0.8 ± 1.3 vs -0.2 ± 1.65; p = 0.031) and fewer documented fasting hypoglycemia episodes (20/44 vs 9/44; p = 0.012), although hepatomegaly frequently persisted. Liver biopsies showed steatosis, fibrosis, and/or chronic hepatitis in 52% of examined cases. A single hepatic adenoma was identified in a 14-year-old male. Overall, the clinical course of GSD IX was favorable, with hepatomegaly, elevated liver enzymes, and dyslipidemia as the most prevalent features. Severe hypoglycemic episodes were uncommon, and no clear genotype-phenotype correlation emerged.
A novel sequence of the PHKG2 mutation associated with the first case of glycogen storage diseases type IXc in Syria: a case report and review of literature.
Glycogen storage diseases are a group of inherited metabolic disorders that affect the body's ability to break down and/or store glycogen. Type IX glycogen storage disease is an inherited disorder caused by a deficiency of phosphorylase kinase, which leads to various symptoms. We report the first reported case in Syria of glycogen storage disease type IXc caused by a novel phosphorylase B kinase catalytic subunit gamma 2 gene mutation, emphasizing the importance of early diagnosis and genetic counseling. A 6-month-old Syrian male infant of Arab ethnicity presented with developmental delay, hepatomegaly, and hypoglycemia. Genetic testing identified a previously unreported phosphorylase B kinase catalytic subunit gamma 2 variant (c.801G > A p.( =)), classified as a variant of uncertain significance. Liver biopsy and clinical features were consistent with glycogen storage disease type IXc. This report expands the current understanding of phosphorylase B kinase catalytic subunit gamma 2-related glycogen storage disease type IXc by documenting a novel synonymous mutation with potential clinical significance. It underscores the critical role of early genetic testing in consanguineous populations, not only for accurate diagnosis but also for guiding family counseling and long-term management. The identification of this novel mutation contributes to expanding the known phosphorylase B kinase catalytic subunit gamma 2 mutation spectrum and stresses the need for genetic counseling in similar populations.
Understanding Glycogen Storage Disease Type IX: A Systematic Review with Clinical Focus-Why It Is Not Benign and Requires Vigilance.
Glycogen storage disease type IX (GSD IX) is a group of inherited metabolic disorders caused by phosphorylase kinase deficiency affecting the liver or muscle. Despite being relatively common among GSDs, GSD IX remains underexplored. A systematic review of GSD IX was conducted per PRISMA guidelines using SCOPUS and PubMed, registered with PROSPERO. Inclusion focused on human clinical studies published up to 31 December 2024. A total of 400 patients with GSD IX were analyzed: 274 IXa (mean age at diagnosis 5.1 years), 72 IXc (mean age at diagnosis 4.9 years), 39 IXb (mean age at diagnosis 4.2 years), and 15 IXd (mean age at diagnosis 44.9 years). Hepatomegaly was commonly reported in types IXa, IXb, and especially IXc (91.7%), but was rare in IXd. Elevated transaminases were frequently observed in types IXa, IXb, and particularly IXc, while uncommon in IXd. Fasting hypoglycemia was occasionally observed in types IXa and IXb, more frequently in IXc (52.7%), and was not reported in IXd. Growth delay or short stature was observed in a substantial proportion of patients with types IXa (43.8%), IXb, and IXc, but was rare in IXd. Muscle involvement was prominent in IXd, with all patients showing elevated CPK (mean 1011 U/L). Neurological involvement was infrequently reported in types IXa and IXc. This systematic review includes the most extensive clinical case history of GSD IX described in the literature. The clinical spectrum of GSD IX varies widely among subtypes, with IXc being the most aggressive. While liver forms are generally present in early childhood, muscle-type IXd shows delayed onset and milder symptoms, often leading to diagnostic delays. For diagnosis, it is essential not to underestimate key clinical features such as hepatic involvement and hypoglycemia in a child under 5 years of age. Other manifestations, including the as-yet unexplored systemic involvement of bone and kidney, remain insufficiently understood and require further investigation. Next-generation sequencing has improved diagnostic precision over traditional biopsy. Dietary management, including uncooked cornstarch, Glycosade®, and high-protein intake, remains the cornerstone of treatment. However, there is a paucity of well-designed, evidence-based studies to determine the most effective therapeutic approach. Despite its historically perceived benign course, the broad phenotypic variability of GSD IX, including progressive liver involvement and potential neurological complications, highlights its substantial clinical relevance and underscores the need for accurate diagnostic classification and long-term multidisciplinary follow-up. Glycogen storage diseases (GSDs) are inherited inborn errors of carbohydrate metabolism that result in abnormal glycogen storage. The onset can range from neonatal life to adulthood, and clinical manifestations result either from a failure to convert glycogen into energy or the toxic accumulation of glycogen. Glycogen is a branched polymer comprised of glucose monomers (see Image. Glycogen, Free Glucose Release, and Glycogen Storage Diseases, Figure 1). After a meal, the plasma glucose level rises, stimulating the storage of the excess in cytoplasmic glycogen. The liver contains the highest percentage of glycogen by weight (about 10%), whereas muscles can store about 2% by weight. Nevertheless, since the total muscle mass is greater than the liver mass, the total mass of glycogen in muscles is about twice that of the liver. When needed, the glycogen polymer can be broken down into glucose monomers and utilized for energy production. Defects in the enzymes and transporters for these processes cause GSDs. An increasing number of GSDs are being identified, but most are very rare. These subtypes are classified numerically in the order of recognition and identification of the enzyme defect causing the disorder. Classification of Glycogen Storage Disorder GSDs that primarily affect the liver include the following: Glycogen synthase-2 deficiency (GSD type 0a). Glucose-6-phosphatase deficiency (GSD type Ia). Glucose-6-phosphate transporter deficiency (GSD type Ib) . Glycogen debrancher deficiency (GSD type III) . Glycogen branching enzyme deficiency (GSD type IV) . Liver phosphorylase deficiency (GSD type VI) . Phosphorylase kinase deficiency (GSD type IXa). GLUT2 deficiency or Fanconi-Bickel disease. GSDs that primarily affect the skeletal muscles include the following: Muscle phosphorylase deficiency (GSD type V). Phosphofructokinase deficiency (GSD type VII). Phosphoglycerate mutase deficiency (GSD type X). Lactate dehydrogenase A deficiency (GSD type XI) . Aldolase A deficiency (GSD type XII); β-enolase deficiency (GSD type XIII). Phosphoglucomutase-1 deficiency (GSD type XIV). GSDs that affect both skeletal and cardiac muscles include the following: Lysosomal acid maltase deficiency (GSD type IIa). Lysosome-associated membrane protein 2 deficiency (GSD type IIb). Glycogenin-1 deficiency (GSD type XV). Muscle glycogen synthase deficiency (GSD type 0b).
Exercise Intolerance in McArdle Disease: A Role for Cardiac Impairment? A Preliminary Study in Humans and Mice.
Whether cardiac impairment can be fully discarded in McArdle disease-the paradigm of "exercise intolerance," caused by inherited deficiency of the skeletal muscle-specific glycogen phosphorylase isoform ("myophosphorylase")-remains to be determined. Eight patients with McArdle disease and seven age/sex-matched controls performed a 15-min moderate, constant-load cycle-ergometer exercise bout followed by a maximal ramp test. Electrocardiographic and two-dimensional transthoracic (for cardiac dimension's assessment) and speckle tracking (for left ventricular global longitudinal strain (GLS) assessments) echocardiographic evaluations were performed at baseline. Electrocardiographic and GLS assessments were also performed during constant-load exercise and immediately upon maximal exertion. Four human heart biopsies were obtained in individuals without McArdle disease, and in-depth histological/molecular analyses were performed in McArdle and wild-type mouse hearts. Exercise intolerance was confirmed in patients ("second wind" during constant-load exercise, -55% peak power output vs controls). As opposed to controls, patients showed a decrease in GLS during constant-load exercise, especially upon second wind occurrence, but with no other between-group difference in cardiac structure/function. Human cardiac biopsies showed that all three glycogen phosphorylase-myophosphorylase, but also liver and especially brain-isoforms are expressed in the normal adult heart, thereby theoretically compensating for eventual myophosphorylase deficiency. No overall histological (including glycogen depots), cytoskeleton, metabolic, or mitochondrial (morphology/network/distribution) differences were found between McArdle and wild-type mouse hearts, except for lower levels of pyruvate kinase M2 and translocase of outer-membrane 20-kDa subunit in the former. This study provides preliminary evidence that cardiac structure and function seem to be preserved in patients with McArdle disease. However, the role for an impaired cardiac contractility associated with the second wind phenomenon should be further explored.
Publicações recentes
Hepatic Glycogen Storage Diseases in Brazil: A Multicenter Study.
Glycogen Storage Disease Type VI.
Sodium taurocholate cotransporter polypeptide deficiency combined with novel PYGL mutations in glycogen storage disease type VI: a rare case report.
Glycogen storage disease type IX: Long-term follow-up of 52 patients from three European countries.
Glutathione S-transferase Mu 3 Mitigates Alcohol-induced Hepatic Lipid Dysregulation via PYGM Suppression.
📚 EuropePMCmostrando 33
Sodium taurocholate cotransporter polypeptide deficiency combined with novel PYGL mutations in glycogen storage disease type VI: a rare case report.
Clinics and research in hepatology and gastroenterologyGlycogen storage disease type IX: Long-term follow-up of 52 patients from three European countries.
Molecular genetics and metabolism reportsA novel sequence of the PHKG2 mutation associated with the first case of glycogen storage diseases type IXc in Syria: a case report and review of literature.
Journal of medical case reportsUnderstanding Glycogen Storage Disease Type IX: A Systematic Review with Clinical Focus-Why It Is Not Benign and Requires Vigilance.
GenesProgressive liver disease and dysregulated glycogen metabolism in murine GSD IX γ2 models human disease.
Molecular genetics and metabolismThe Autophagic Activator GHF-201 Can Alleviate Pathology in a Mouse Model and in Patient Fibroblasts of Type III Glycogenosis.
BiomoleculesExercise Intolerance in McArdle Disease: A Role for Cardiac Impairment? A Preliminary Study in Humans and Mice.
Medicine and science in sports and exerciseA Functional Human Glycogen Debranching Enzyme Encoded by a Synthetic Gene: Its Implications for Glycogen Storage Disease Type III Management.
Protein and peptide lettersReport of an Iranian child with chronic abdominal pain and constipation diagnosed as glycogen storage disease type IX: a case report.
Journal of medical case reports[Splicing abnormalities caused by a novel mutation in the PHKA2 gene in children with glycogen storage disease type IX].
Zhonghua gan zang bing za zhi = Zhonghua ganzangbing zazhi = Chinese journal of hepatologyGlycogen Storage Disease Phenotypes Accompanying the Perturbation of the Methionine Cycle in NDRG3-Deficient Mouse Livers.
CellsGeneration of human induced pluripotent stem cell line, KRIBBi003-A, from urinary cells of a patient with glycogen storage disease type IXa.
Stem cell researchIdentification of a novel mutation in the PHKA2 gene in a child with liver cirrhosis.
Journal of pediatric endocrinology & metabolism : JPEMGlycogen accumulation and phase separation drives liver tumor initiation.
CellThe Phenotypic and Genetic Spectrum of Glycogen Storage Disease Type VI.
GenesProfound neonatal lactic acidosis and renal tubulopathy in a patient with glycogen storage disease type IXɑ2 secondary to a de novo pathogenic variant in PHKA2.
Molecular genetics and metabolism reportsCharacterization of liver GSD IX γ2 pathophysiology in a novel Phkg2-/- mouse model.
Molecular genetics and metabolismGlycogen storage disease type VI with a novel PYGL mutation: Two case reports and literature review.
MedicineNovel mutations in the PHKB gene in an iranian girl with severe liver involvement and glycogen storage disease type IX: a case report and review of literature.
BMC pediatricsBenign or not benign? Deep phenotyping of liver Glycogen Storage Disease IX.
Molecular genetics and metabolismVariability of clinical and biochemical phenotype in liver phosphorylase kinase deficiency with variants in the phosphorylase kinase (PHKG2) gene.
Journal of pediatric endocrinology & metabolism : JPEMLiver histology in children with glycogen storage disorders type VI and IX.
Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the LiverNovel PYGL mutations in Chinese children leading to glycogen storage disease type VI: two case reports.
BMC medical geneticsMolecular diagnosis of glycogen storage disease type IX using a glycogen storage disease gene panel.
European journal of medical geneticsGlycogen storage disease type VI: clinical course and molecular background.
European journal of pediatricsLiver Glycogen Phosphorylase Deficiency Leads to Profibrogenic Phenotype in a Murine Model of Glycogen Storage Disease Type VI.
Hepatology communicationsA novel PHKA2 mutation in a Chinese child with glycogen storage disease type IXa: a case report and literature review.
BMC medical geneticsDiagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG).
Genetics in medicine : official journal of the American College of Medical GeneticsPHKG2 mutation spectrum in glycogen storage disease type IXc: a case report and review of the literature.
Journal of pediatric endocrinology & metabolism : JPEMHepatocytes contribute to residual glucose production in a mouse model for glycogen storage disease type Ia.
Hepatology (Baltimore, Md.)Clinical and Molecular Variability in Patients with PHKA2 Variants and Liver Phosphorylase b Kinase Deficiency.
JIMD reportsDifferential Muscle Involvement in Mice and Humans Affected by McArdle Disease.
Journal of neuropathology and experimental neurologySodium valproate increases the brain isoform of glycogen phosphorylase: looking for a compensation mechanism in McArdle disease using a mouse primary skeletal-muscle culture in vitro.
Disease models & mechanismsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença de armazenamento de glicogênio por deficiência de glicogenofosforilase hepática.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença de armazenamento de glicogênio por deficiência de glicogenofosforilase hepática
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Sodium taurocholate cotransporter polypeptide deficiency combined with novel PYGL mutations in glycogen storage disease type VI: a rare case report.
- Glycogen storage disease type IX: Long-term follow-up of 52 patients from three European countries.
- A novel sequence of the PHKG2 mutation associated with the first case of glycogen storage diseases type IXc in Syria: a case report and review of literature.
- Understanding Glycogen Storage Disease Type IX: A Systematic Review with Clinical Focus-Why It Is Not Benign and Requires Vigilance.
- Exercise Intolerance in McArdle Disease: A Role for Cardiac Impairment? A Preliminary Study in Humans and Mice.
- Hepatic Glycogen Storage Diseases in Brazil: A Multicenter Study.
- Glycogen Storage Disease Type VI.
- Glutathione S-transferase Mu 3 Mitigates Alcohol-induced Hepatic Lipid Dysregulation via PYGM Suppression.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:369(Orphanet)
- OMIM OMIM:232700(OMIM)
- MONDO:0009294(MONDO)
- GARD:6529(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1947298(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
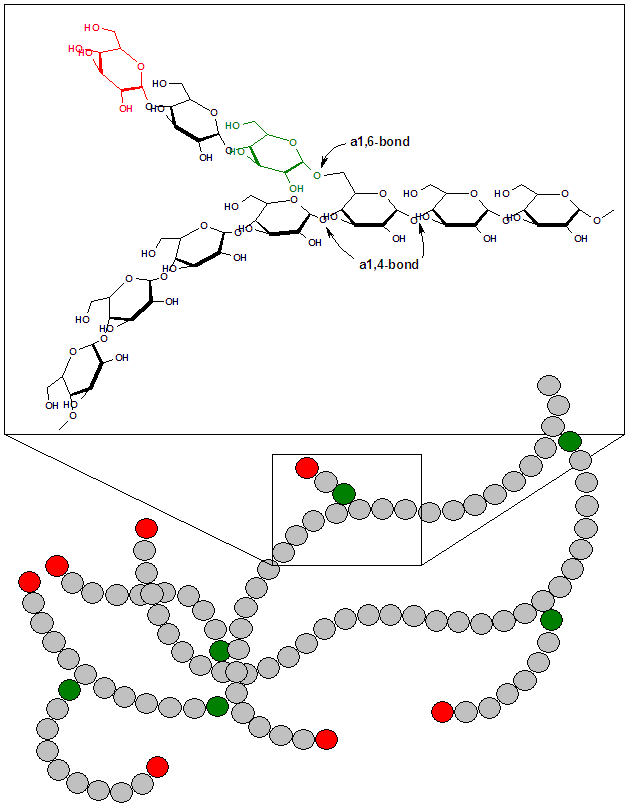
Doença de armazenamento de glicogênio por deficiência de glicogenofosforilase hepática
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata