A doença de Camurati-Englemann (CED) é uma síndrome de displasia óssea rara e clinicamente variável, caracterizada por hiperostose dos ossos longos, crânio, coluna e pelve, associada a dor intensa nas extremidades, marcha gingada de base ampla, contraturas articulares, fraqueza muscular e fadiga fácil.
Introdução
O que você precisa saber de cara
A doença de Camurati-Englemann (CED) é uma síndrome de displasia óssea rara e clinicamente variável, caracterizada por hiperostose dos ossos longos, crânio, coluna e pelve, associada a dor intensa nas extremidades, marcha gingada de base ampla, contraturas articulares, fraqueza muscular e fadiga fácil.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 27 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 71 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisTransforming growth factor beta-1 proprotein: Precursor of the Latency-associated peptide (LAP) and Transforming growth factor beta-1 (TGF-beta-1) chains, which constitute the regulatory and active subunit of TGF-beta-1, respectively Required to maintain the Transforming growth factor beta-1 (TGF-beta-1) chain in a latent state during storage in extracellular matrix (PubMed:28117447). Associates non-covalently with TGF-beta-1 and regulates its activation via interaction with 'milieu molecules',
Secreted, extracellular space, extracellular matrixSecreted
Camurati-Engelmann disease
An autosomal dominant disorder characterized by hyperostosis and sclerosis of the diaphyses of long bones. The disease typically presents in early childhood with pain, muscular weakness and waddling gait, and in some cases other features such as exophthalmos, facial paralysis, hearing difficulties and loss of vision.
Variantes genéticas (ClinVar)
39 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 4 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
21 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença de Camurati-Engelmann
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
Rare diseases, defined by the 2002 Rare Disease Act, affect fewer than 5 in 10,000 individuals. Rare metabolic bone diseases (MBDs), such as osteogenesis imperfecta, hypophosphatasia, osteopetrosis, and other unclassified disorders, can disrupt bone development and remodeling, posing diagnostic and management challenges. This study analyzed data from the rarembd.in registry (2010-2024), a 15-year database documenting only rare MBDs. Clinical presentation and demographic data of patients with rare MBDs were collated. Common MBDs (osteoporosis, primary hyperparathyroidism) were excluded. Genetic testing was performed in a subset of patients. There was a total of 218 patients with an almost equal gender distribution (male-to-female ratio of 1:1.07) and a mean age of 29.1 ± 18.9 years. The registry identified 29 rare MBDs with three main disease categories: demineralization disorders (50.4%), disorders of bone matrix and cartilage formation (32.5%), and sclerotic disorders (13.7%); with a smaller proportion categorized as unclassified bone disorders (2.7%). Rickets/osteomalacia (27.1%) was the most common, followed by osteogenesis imperfecta (23.4%) and fibrous dysplasia/McCune-Albright syndrome (18.8%). Fractures affected 57.7% of patients, with 24.5% experiencing multiple fractures, while 31.1% exhibited skeletal deformities. Mutation analysis in our registry identified pathogenic variants in the SOST, TGFβ1, SLC34A3, ALPL, and VCP genes, confirming the genetic basis of sclerosteosis, Camurati-Engelmann disease, hypophosphatemic rickets, hypophosphatasia, and IBMPFD, respectively. Different management strategies were used that included teriparatide, bisphosphonates (zoledronate or alendronate) with total contact casting, intralesional zoledronate, denosumab, calcium, active vitamin D, and recombinant human growth hormone. Total parathyroidectomy was performed in specific cases. The registry classified RMBDs into four categories, with demineralization disorders being the most common, followed by bone matrix/cartilage formation disorders, sclerotic diseases, and unclassified cases. There were 29 RMBDs, and rickets/osteomalacia was the most prevalent subtype, tumor-induced osteomalacia followed by familial hypophosphatemic osteomalacia. Among the unclassified bone disorders, fragility fractures emerged as the most common presentation.
Health-related quality of life in patients with Camurati-Engelmann disease: A case series study.
Camurati-Engelmann disease (CED) is an extremely rare autosomal dominant bone dysplasia characterized by hyperostosis of the long bones and, in severe cases, of the skull and axial skeleton. This study aims to analyze the impact on quality of life in a non-selected group of CED patients. This monocentric case series study included 15 patients who completed a survey that collected demographic characteristics, disease-related information and a validated SF-12 questionnaire to assess quality of life. Physical (PCS-12) and Mental (MCS-12) Component Summary scores were compared with the mean SF-12 scores of the U.S. reference population. Patients had significantly lower PCS-12 score [29.8 (7.5)] compared to the reference population [50.0 (10.0)] (p<0.0001). However, there were no differences in the MCS-12 score [48.8 (11.23) vs 50.0 (10.0)]. The most reported symptoms were fatigue (100%), pain in the limbs (93.3%) and muscle pain (86.7%). CED patients have significantly lower physical quality of life than the general population, due to the high prevalence of physically limiting problems. However, mental health appears unaffected.
Transforming growth factor, beta-2 gene mutation causes autosomal dominant Camurati-Engelmann disease, type 2 (OMIM % 606631).
Camurati-Engelmann disease, type 1 (CED1, OMIM # 131300) is the rare autosomal dominant skeletal dysplasia caused by select heterozygous loss-of-function defects within the gene TGFB1, which encodes transforming growth factor beta 1 (TGFB1). CED1 mutations are found in TGFB1 exons 1-4 that form the latency-associated peptide (LAP) of pro-TGFB1. Consequently, skeletal action of TGFB1 increases and thereby enhances bone formation manifest clinically as "progressive diaphyseal dysplasia". Beginning 24 years ago negative TGFB1 analysis suggested rare genetic heterogeneity for CED, and Online Mendelian Inheritance In Man designated, of unknown etiology, "CED2" (OMIM % 606631). In 2024, three sporadic occurrences considered CED2 were reported to harbor either of two mutations of TGFB2, which encodes the LAP of transforming growth factor beta 2 (TGFB2). Herein, three adults (father, son, daughter) having the CED2 phenotype in a Peruvian family revealed a novel missense variant (c.108G > T, p.R36S) within the TGFB2 LAP domain. Debilitating painful skeletal disease featuring hyperostosis of entire long bones, worse in the men, presented early in childhood. Aminobisphosphonate therapy seemed helpful. Their TGFB2 variant was within a highly conserved domain across species, absent in the gnomAD database, "possibly damaging" by Polyphen-2, not tolerated by SIFT, homologous with TGFB1 at the same amino acid position (R36) as one reported TGFB2 mutation, co-segregated as autosomal dominant, and "likely pathogenic" per ACMG guidelines.
Insights into Natural History, Phenotypic, and Molecular Spectrum in a Large Cohort of Osteosclerotic Disorders.
Osteosclerotic bone diseases include more than 30 rare diseases characterized by excessive bone formation. The aim of this study is to compare the molecular pathogenesis and prognostic features of 12 different osteosclerotic diseases. Thirty-four patients from 23 families were included, 25 of whom were followed for a period of one to 22 years. Exome sequencing was performed in 20 families. Primary hypertrophic osteoarthropathy (PHOAR1/2) was found in 12 patients, followed by juvenile Paget's disease (JPD)-5 in five, craniometaphyseal dysplasia (CMD) and Camurati-Engelmann disease (CED) in four, Ghosal hematodiaphyseal dysplasia (GHDD) in three patients, sclerosteosis-1 in two patients, and ultra-rare diseases including trichothiodystrophy-1, prenatal Caffey disease, melorheosteosis, and Lenz-Majewski hyperostotic dwarfism in one patient each. Patients with CMD and sclerosteosis-1 had severe cranial sclerosis leading to facial dysmorphism. CMD was characterized by metaphyseal widening, radiolucency, and diaphyseal sclerosis of the long bones in early childhood and later developed Erlenmeyer flask deformity sparing the vertebrae and pelvis, whereas sclerosteosis-1 manifested as generalized sclerosis. CED and GHDD share bone pain, difficulty in walking, and diaphyseal sclerosis, with some patients also having bone marrow involvement. Interestingly, patients with CED and JPD-5 showed osteopenia in early childhood, followed by the development of osteosclerosis in late childhood. Clinical and radiologic findings improved over time in PHOAR1 patients, whereas they progressed in JPD-5 and trichothiodystrophy-1 patients. Intra- and interfamilial clinical differences were observed in CMD, CED, JPD-5, and GHDD. The knowledge gained about the natural history of osteosclerotic diseases will make an important contribution to their diagnosis and management.
Identification of a novel TGFB1 variant in a patient with Camurati-Engelmann disease responsive to alendronate.
Publicações recentes
Camurati-Engelmann disease with bilateral proptosis and optic neuropathy: a case report and literature review.
Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
Identification of a novel TGFB1 variant in a patient with Camurati-Engelmann disease responsive to alendronate.
Corrigendum to "Transforming growth factor, beta-2 gene mutation causes autosomal dominant Camurati-Engelmann disease, type 2 (OMIM % 606631)" [Bone 197 (2025) 117477].
Health-related quality of life in patients with Camurati-Engelmann disease: A case series study.
📚 EuropePMC155 artigos no totalmostrando 69
Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
Calcified tissue internationalIdentification of a novel TGFB1 variant in a patient with Camurati-Engelmann disease responsive to alendronate.
Rheumatology (Oxford, England)Corrigendum to "Transforming growth factor, beta-2 gene mutation causes autosomal dominant Camurati-Engelmann disease, type 2 (OMIM % 606631)" [Bone 197 (2025) 117477].
BoneHealth-related quality of life in patients with Camurati-Engelmann disease: A case series study.
Medicina clinicaTransforming growth factor, beta-2 gene mutation causes autosomal dominant Camurati-Engelmann disease, type 2 (OMIM % 606631).
BoneInsights into Natural History, Phenotypic, and Molecular Spectrum in a Large Cohort of Osteosclerotic Disorders.
Calcified tissue internationalSclerosing bone dysplasias: a pictorial essay.
Radiologia brasileiraUnilateral hemicraniectomy with titanium cranioplasty for the treatment of high intracranial pressure in a pediatric patient with Camurati-Engelmann disease: illustrative case.
Journal of neurosurgery. Case lessonsPhenotypic Variability in Camurati-Engelmann Disease: A Case Report of a Family with the c.653G>A Pathogenic Variant in the TGFB1 Gene.
GenesUnveiling the uncommon: diagnostic journey of camurati-engelmann disease in a pediatric patient.
Pediatric rheumatology online journalHeterozygous mutations in the straitjacket region of the latency-associated peptide domain of TGFB2 cause Camurati-Engelmann disease type II.
Journal of human geneticsA new case of Melnick-Needles syndrome with skeletal manifestations: A case report.
International journal of surgery case reportsThe Impact of Genetic Variability of TGF-Beta Signaling Biomarkers in Major Craniofacial Syndromes.
Advances in experimental medicine and biologyA case of papilledema in Camurati-Engelmann disease treated effectively with prednisolone.
Clinical pediatric endocrinology : case reports and clinical investigations : official journal of the Japanese Society for Pediatric EndocrinologyCamurati-Engelmann Disease: A Case-Based Review About an Ultrarare Bone Dysplasia.
European journal of rheumatologyDiagnosing Camurati-Engelmann disease-the age of whole-exome sequencing.
Rheumatology (Oxford, England)Clinical characteristics and the influence of rs1800470 in patients with Camurati-Engelmann disease.
Frontiers in endocrinologyAberrant activation of TGF-β1 induces high bone turnover via Rho GTPases-mediated cytoskeletal remodeling in Camurati-Engelmann disease.
Frontiers in endocrinologyCamurati-Engelmann disease combined with transethmoidal meningoencephalocele: illustrative case.
Journal of neurosurgery. Case lessonsImprovement of Bone Health and Initiation of Puberty Development in Camurati-Engelmann Disease With Glucocorticoid and Losartan Treatment: A Case Report and Review of Literature.
Frontiers in endocrinologyLosartan as a Steroid-Sparing Adjunct in a Patient With Features of Refractory Camurati-Engelmann Disease.
AACE clinical case reportsCamurati-Engelmann Disease Complicated by Hypopituitarism: Management Challenges and Literature Review of Outcomes With Bisphosphonates.
AACE clinical case reportsClinical characteristics and identification of a novel TGFB1 variant in three unrelated Chinese families with Camurati-Engelmann disease.
Molecular genetics & genomic medicineLooking for the skeleton in the closet-rare genetic diagnoses in patients with diabetes and skeletal manifestations.
Acta diabetologicaDual Biologic Therapy in an Adolescent With Camurati-Engelmann Disease and Crohn Disease.
JPGN reportsAlteration of Bone Density, Microarchitecture, and Strength in Patients with Camurati-Engelmann Disease: Assessed by HR-pQCT.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchSurgery Treatment of an Adult Patient with Camurati-Engelmann Disease: A Case Report.
JBJS case connectorAnterior Total Hip Arthroplasty With Bulk Femoral Head Autograft in a Patient With Camurati-Engelmann Disease.
Arthroplasty todaySignificant Improvement After Surgery for a Symptomatic Osteoblastoma in a Patient with Camurati-Engelmann Disease: Case Report and Literature Review.
Calcified tissue internationalDecompression of the internal auditory canal via the retrosigmoid approach in a patient with Camurati-Engelmann disease: illustrative case.
Journal of neurosurgery. Case lessonsCamurati-Engelmann disease or Ribbing disease.
Joint bone spineLooking for new anabolic treatment from rare diseases of bone formation.
The Journal of endocrinologyA new LRP6 variant and Camurati-Engelmann-like disease.
BoneEfficacy of denosumab therapy after alendronate treatment for a 66-year-old woman with Camurati-Engelmann disease and osteoporosis: a case report.
Modern rheumatology case reportsA patient with Camurati-Engelmann disease presenting bilateral TMJ ankylosis: A case report.
International journal of surgery case reportsA Case Report of a 44-Year-Old Woman With Camurati-Englemann Disease: A Case Report.
JBJS case connectorWhole-Body Magnetic Resonance Imaging in Camurati-Engelmann Disease.
JBMR plusGenetic testing is useful in adults with limited phenotypes of genetic skeletal conditions.
BoneCamurati-Engelmann Disease with Good Treatment Response to Losartan.
Indian journal of nuclear medicine : IJNM : the official journal of the Society of Nuclear Medicine, IndiaSeropositive Rheumatoid Arthritis with Very Unusual X-ray Findings.
European journal of case reports in internal medicineCamurati-Engelmann Disease.
Calcified tissue internationalObservations on the Natural History of Camurati-Engelmann Disease.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchAberrant activation of latent transforming growth factor-β initiates the onset of temporomandibular joint osteoarthritis.
Bone researchBone-targeted delivery of TGF-β type 1 receptor inhibitor rescues uncoupled bone remodeling in Camurati-Engelmann disease.
Annals of the New York Academy of SciencesCamurati-Engelmann disease: a case report from sub-Saharan Africa.
Oxford medical case reportsTotal Hip Arthroplasty in a Patient with Camurati-Engelmann Disease: A Case Report.
JBJS case connectorClinical characteristics and treatment outcomes in Camurati-Engelmann disease: A case series.
MedicineRegarding Camurati-Engelmann Disease: In Reply.
Clinics in orthopedic surgeryRegarding Camurati-Engelmann Disease: To the Editor.
Clinics in orthopedic surgeryFailure of conventional treatment and losartan in Camurati-Engelmann disease: A case report.
Joint bone spineSignificant Improvement of Clinical Symptoms, Bone Lesions, and Bone Turnover after Long-Term Zoledronic Acid Treatment in Patients with a Severe Form of Camurati-Engelmann Disease.
Molecular syndromologyPain improvement in Camurati-Engelmann disease after anti-TNFα therapy.
BMJ case reportsCamurati-Engelmann disease: New clinical insights in an Egyptian case report.
Journal of orthopaedic science : official journal of the Japanese Orthopaedic AssociationDiscrepancy between bone density and bone material strength index in three siblings with Camurati-Engelmann disease.
Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USAAngioid streaks in a case of Camurati-Engelmann disease.
Indian journal of ophthalmologyCamurati-Engelmann disease-a rare cause of tetany identified on bone scintigraphy: A case report.
MedicineA Rare Sporadic Case of Camurati-Engelmann Disease With Jaw Involvement.
Journal of oral and maxillofacial surgery : official journal of the American Association of Oral and Maxillofacial SurgeonsOrthopedic Manifestations of Type I Camurati-Engelmann Disease.
Clinics in orthopedic surgeryRepeat Intracranial Expansion After Skull Regrowth in Hyperostotic Disease: Technical Note.
World neurosurgeryImaging aspects of Camurati-Engelmann disease.
Revista da Associacao Medica Brasileira (1992)Clinical diagnosis and mutation analysis of a Chinese family with Camurati-Engelmann disease.
Molecular medicine reportsAn ENU-induced p.C225S missense mutation in the mouse Tgfb1 gene does not cause Camurati-Engelmann disease-like skeletal phenotypes.
Experimental animalsMild Camurati‑Engelamann disease presenting with exophthalmos as the first and only manifestation: A case report.
Molecular medicine reports[Camurati-Engelmann disease].
Nihon rinsho. Japanese journal of clinical medicineExcess TGF-β mediates muscle weakness associated with bone metastases in mice.
Nature medicineReduced Dentin Matrix Protein Expression in Camurati-Engelmann Disease Transgenic Mouse Model.
Journal of cellular physiologyBilateral papilloedema in Camurati-Engelmann disease.
BMJ case reportsSkeletal Dysplasia Presenting as a Neuromuscular Disorder - Report of a Family with Camurati-Engelmann Syndrome.
MaedicaCorrelative bone imaging in a case of Schnitzler's syndrome and brief review of the literature.
Hellenic journal of nuclear medicineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença de Camurati-Engelmann.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença de Camurati-Engelmann
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
- Health-related quality of life in patients with Camurati-Engelmann disease: A case series study.
- Transforming growth factor, beta-2 gene mutation causes autosomal dominant Camurati-Engelmann disease, type 2 (OMIM % 606631).
- Insights into Natural History, Phenotypic, and Molecular Spectrum in a Large Cohort of Osteosclerotic Disorders.
- Identification of a novel TGFB1 variant in a patient with Camurati-Engelmann disease responsive to alendronate.
- Camurati-Engelmann disease with bilateral proptosis and optic neuropathy: a case report and literature review.
- Corrigendum to "Transforming growth factor, beta-2 gene mutation causes autosomal dominant Camurati-Engelmann disease, type 2 (OMIM % 606631)" [Bone 197 (2025) 117477].
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1328(Orphanet)
- OMIM OMIM:131300(OMIM)
- MONDO:0007542(MONDO)
- GARD:1072(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q498487(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
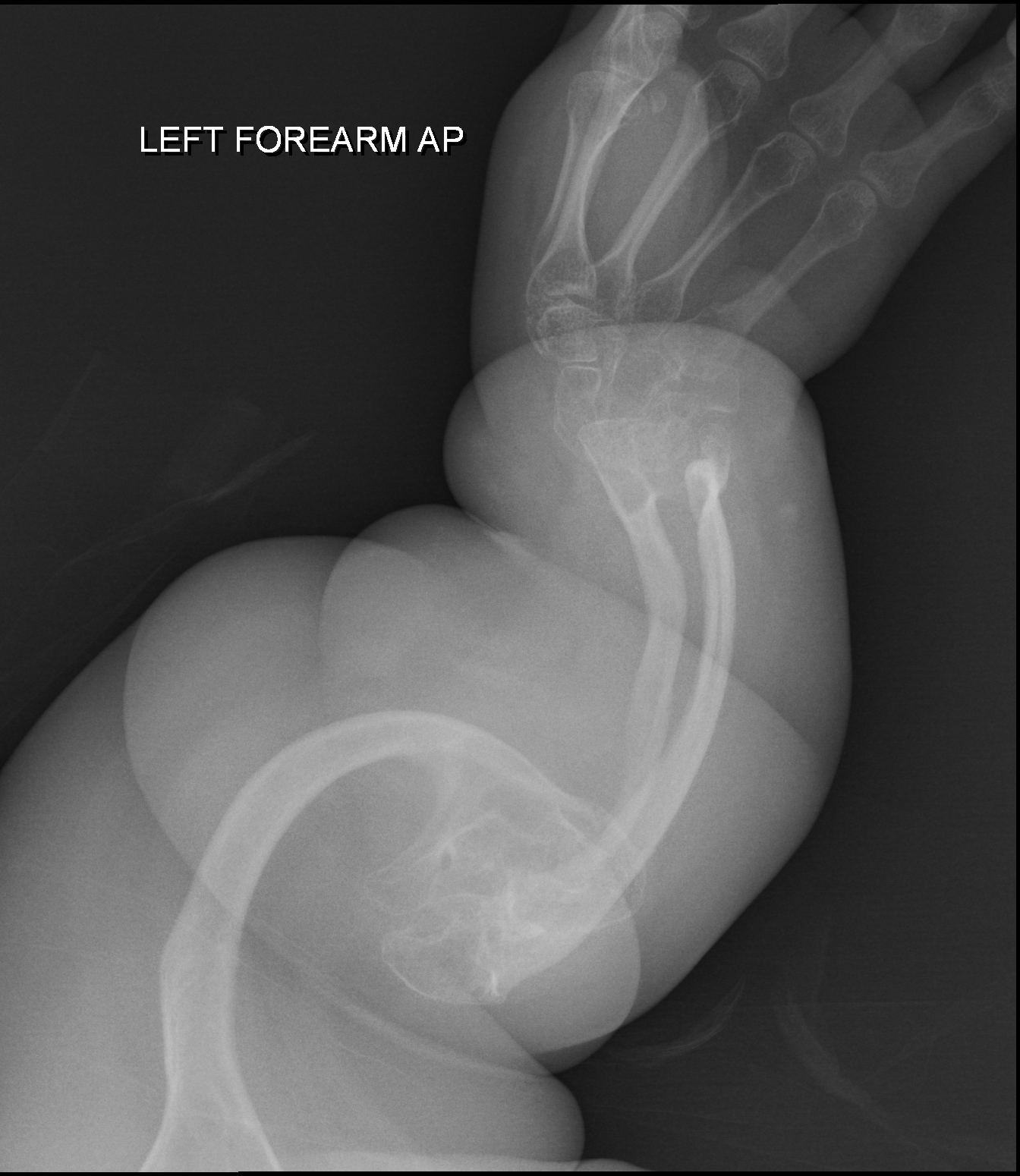
Doença de Camurati-Engelmann
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata