Síndrome de Cockayne causada por mutação(ões) no gene ERCC8, que codifica a proteína de reparo por excisão de DNA ERCC-8.
Introdução
O que você precisa saber de cara
Síndrome de Cockayne causada por mutação(ões) no gene ERCC8, que codifica a proteína de reparo por excisão de DNA ERCC-8.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 36 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 117 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição.
Curadoria gene-doença
fontes oficiaisEssential factor involved in transcription-coupled nucleotide excision repair (TC-NER), a process during which RNA polymerase II-blocking lesions are rapidly removed from the transcribed strand of active genes (PubMed:16246722, PubMed:20541997, PubMed:22483866, PubMed:26620705, PubMed:32355176, PubMed:34526721, PubMed:38316879, PubMed:38600235, PubMed:38600236). Plays a central role in the initiation of the TC-NER process: specifically recognizes and binds RNA polymerase II stalled at a lesion,
NucleusChromosome
Cockayne syndrome B
A rare disorder characterized by cutaneous sensitivity to sunlight, abnormal and slow growth, cachectic dwarfism, progeroid appearance, progressive pigmentary retinopathy and sensorineural deafness. There is delayed neural development and severe progressive neurologic degeneration resulting in intellectual disability. Two clinical forms are recognized: in the classical form or Cockayne syndrome type 1, the symptoms are progressive and typically become apparent within the first few years or life; the less common Cockayne syndrome type 2 is characterized by more severe symptoms that manifest prenatally. Cockayne syndrome shows some overlap with certain forms of xeroderma pigmentosum. Unlike xeroderma pigmentosum, patients with Cockayne syndrome do not manifest increased freckling and other pigmentation abnormalities in the skin and have no significant increase in skin cancer.
Catalytic component of a structure-specific DNA repair endonuclease responsible for the 5-prime incision during DNA repair, and which is essential for nucleotide excision repair (NER) and interstrand cross-link (ICL) repair
NucleusChromosome
Xeroderma pigmentosum complementation group F
An autosomal recessive pigmentary skin disorder characterized by solar hypersensitivity of the skin, high predisposition for developing cancers on areas exposed to sunlight and, in some cases, neurological abnormalities. The skin develops marked freckling and other pigmentation abnormalities. XP-F patients show a mild phenotype.
Substrate-recognition component of the CSA complex, a DCX (DDB1-CUL4-X-box) E3 ubiquitin-protein ligase complex, involved in transcription-coupled nucleotide excision repair (TC-NER), a process during which RNA polymerase II-blocking lesions are rapidly removed from the transcribed strand of active genes (PubMed:12732143, PubMed:16751180, PubMed:16964240, PubMed:32142649, PubMed:34526721, PubMed:38316879, PubMed:38600235, PubMed:38600236). Following recruitment to lesion-stalled RNA polymerase I
NucleusChromosomeNucleus matrix
Cockayne syndrome A
A rare disorder characterized by cutaneous sensitivity to sunlight, abnormal and slow growth, cachectic dwarfism, progeroid appearance, progressive pigmentary retinopathy and sensorineural deafness. There is delayed neural development and severe progressive neurologic degeneration resulting in intellectual disability. Two clinical forms are recognized: in the classical form or Cockayne syndrome type 1, the symptoms are progressive and typically become apparent within the first few years or life; the less common Cockayne syndrome type 2 is characterized by more severe symptoms that manifest prenatally. Cockayne syndrome shows some overlap with certain forms of xeroderma pigmentosum. Unlike xeroderma pigmentosum, patients with Cockayne syndrome do not manifest increased freckling and other pigmentation abnormalities in the skin and have no significant increase in skin cancer.
Variantes genéticas (ClinVar)
855 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 146 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
12 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Cockayne tipo 1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
1 ensaios clínicos encontrados.
Publicações mais relevantes
Publicações recentes
Ver todas no PubMed📚 EuropePMC606 artigos no totalmostrando 1
Ver todos os 606 no EuropePMCAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Cockayne tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Cockayne tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:90321(Orphanet)
- OMIM OMIM:216400(OMIM)
- MONDO:0019569(MONDO)
- GARD:1415(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q914389(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
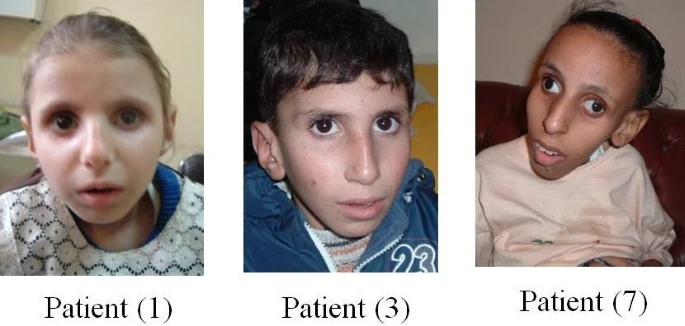
Síndrome Cockayne tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata