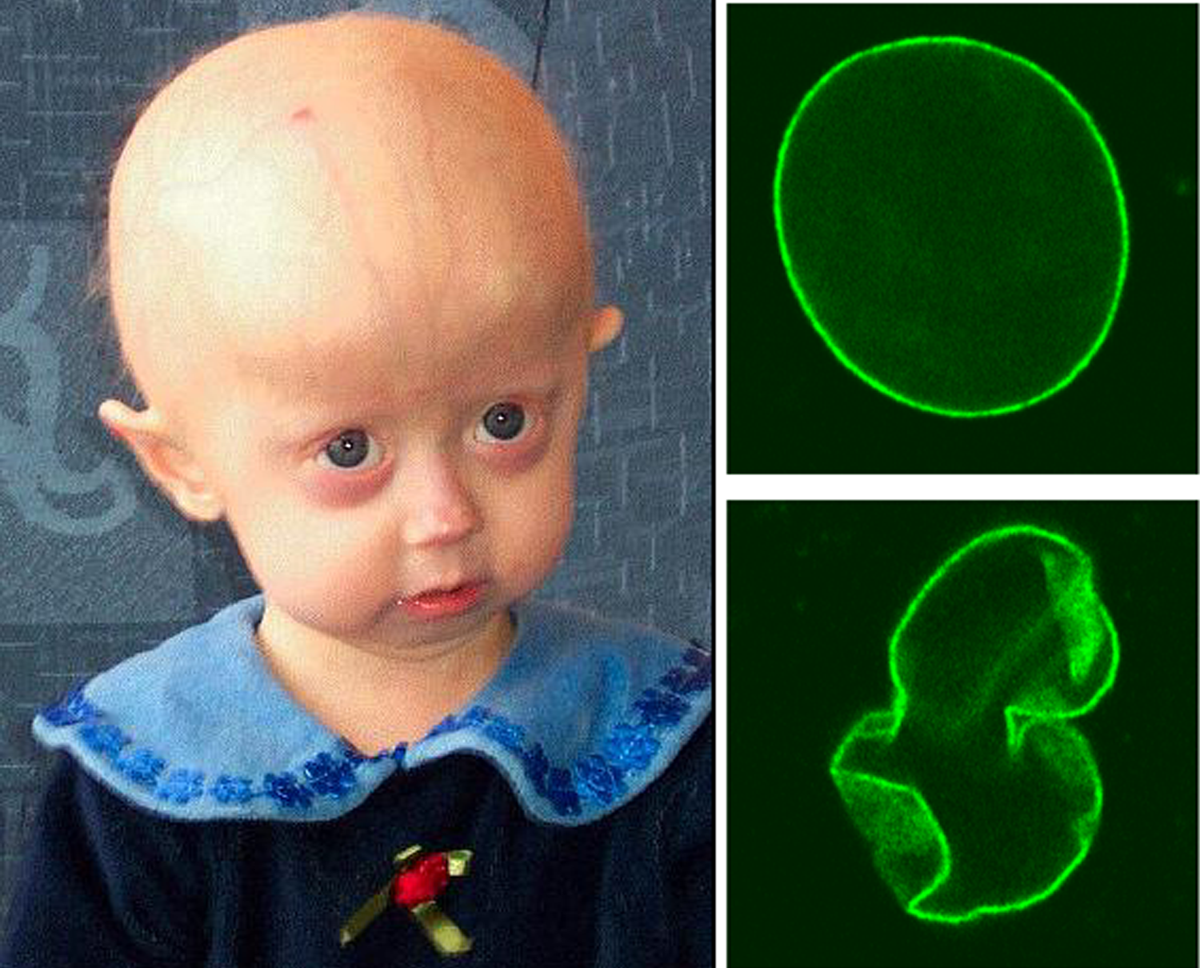
A síndrome semelhante à Síndrome de Quebra de Nijmegen é uma doença genética rara que causa múltiplas alterações congênitas (presentes desde o nascimento) e características físicas incomuns. É caracterizada por atraso no crescimento, baixa estatura, atraso no desenvolvimento e deficiência intelectual. Pessoas com a síndrome também podem apresentar alterações no crânio e no rosto, como microcefalia grave (cabeça menor que o normal), testa inclinada, olhos proeminentes (saltados), ponte nasal larga (a parte de cima do nariz é mais larga), septo nasal subdesenvolvido (a parede que divide as narinas é pequena) e pregas epicânticas (dobras de pele nos cantos internos dos olhos). Outras características incluem instabilidade espontânea dos cromossomos (eles se quebram com facilidade), células muito sensíveis à radiação ionizante (como a de raios-X) e uma capacidade do DNA de se replicar mesmo na presença de radiação, sem que haja infecções graves, problemas no sistema imunológico ou um risco maior de desenvolver câncer. Outras características relatadas incluem espasticidade leve (rigidez muscular), ataxia leve e não progressiva (dificuldade de coordenação que não piora), hipermetropia (dificuldade para enxergar de perto), múltiplos nevos pigmentados (várias pintas ou marcas de nascença), mamilos mais espaçados e clinodactilia (dedos curvados, geralmente o mindinho).
Introdução
O que você precisa saber de cara
A síndrome semelhante à Síndrome de Quebra de Nijmegen é uma doença genética rara que causa múltiplas alterações congênitas (presentes desde o nascimento) e características físicas incomuns. É caracterizada por atraso no crescimento, baixa estatura, atraso no desenvolvimento e deficiência intelectual. Pessoas com a síndrome também podem apresentar alterações no crânio e no rosto, como microcefalia grave (cabeça menor que o normal), testa inclinada, olhos proeminentes (saltados), ponte nasal larga (a parte de cima do nariz é mais larga), septo nasal subdesenvolvido (a parede que divide as narinas é pequena) e pregas epicânticas (dobras de pele nos cantos internos dos olhos). Outras características incluem instabilidade espontânea dos cromossomos (eles se quebram com facilidade), células muito sensíveis à radiação ionizante (como a de raios-X) e uma capacidade do DNA de se replicar mesmo na presença de radiação, sem que haja infecções graves, problemas no sistema imunológico ou um risco maior de desenvolver câncer. Outras características relatadas incluem espasticidade leve (rigidez muscular), ataxia leve e não progressiva (dificuldade de coordenação que não piora), hipermetropia (dificuldade para enxergar de perto), múltiplos nevos pigmentados (várias pintas ou marcas de nascença), mamilos mais espaçados e clinodactilia (dedos curvados, geralmente o mindinho).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 5 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 11 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisComponent of the MRN complex, which plays a central role in double-strand break (DSB) repair, DNA recombination, maintenance of telomere integrity and meiosis (PubMed:15064416, PubMed:21757780, PubMed:27889449, PubMed:28134932, PubMed:28867292, PubMed:9590181, PubMed:9651580, PubMed:9705271). The MRN complex is involved in the repair of DNA double-strand breaks (DSBs) via homologous recombination (HR), an error-free mechanism which primarily occurs during S and G2 phases (PubMed:15064416, PubMed
NucleusChromosome, telomereChromosome
Nijmegen breakage syndrome-like disorder
A disorder similar to Nijmegen breakage syndrome and characterized by chromosomal instability, radiation sensitivity, microcephaly, growth retardation, short stature and bird-like face. Immunodeficiency is absent.
Core component of the MRN complex, which plays a central role in double-strand break (DSB) repair, DNA recombination, maintenance of telomere integrity and meiosis (PubMed:11741547, PubMed:14657032, PubMed:22078559, PubMed:23080121, PubMed:24316220, PubMed:26240375, PubMed:27889449, PubMed:28867292, PubMed:29670289, PubMed:30464262, PubMed:30612738, PubMed:31353207, PubMed:37696958, PubMed:38128537, PubMed:9590181, PubMed:9651580, PubMed:9705271). The MRN complex is involved in the repair of DNA
NucleusChromosomeChromosome, telomere
Ataxia-telangiectasia-like disorder 1
A rare disorder characterized by progressive cerebellar ataxia, dysarthria, abnormal eye movements, and absence of telangiectasia. ATLD patients show normal levels of total IgG, IgA and IgM, although there may be reduced levels of specific functional antibodies. At the cellular level, ATLD exhibits hypersensitivity to ionizing radiation and radioresistant DNA synthesis.
Variantes genéticas (ClinVar)
1.048 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 415 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
22 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença síndrome de quebras de Nijmegen-like
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Novel RAD50 variants lead to Nijmegen Breakage Syndrome-like disorder and unplanned recombinant human growth hormone treatment response.
Human RAD50 gene mutations cause Nijmegen Breakage Syndrome-like disease, characterized by severe prenatal and postpartum growth retardation and microcephaly. It is very rare (less than 5 cases) with limited clinical data and treatment experience. Clinical information was collected on a boy with microcephaly and severe growth restriction, including birth history, clinical features, unplanned response to recombinant human growth hormone treatment, and five-year follow-up after growth hormone discontinuation. The child underwent trio-based whole-exome sequencing and Sanger sequencing to validate the mutation. Constructed variant plasmids were used for in vitro functional experiments and Western blots to evaluate the potential impact of the variants. The boy was born at full term, with substantial growth retardation from infancy to early childhood. At the age of 4.5 years, the child with syndromic short stature was prescribed recombinant human growth hormone for height correction by junior resident physicians, with no genetic evaluation performed prior to treatment. After 5 years and 9 months of recombinant human growth hormone treatment, genetic analysis was done due to his evident microcephaly and distinctive facial features. Two novel variants (p.His1269Argfs2 and p.Ser844Asn) were identified in the RAD50 gene. Western blotting revealed the presence of the Flag-tag and EGFP in RAD50-wt, but not in RAD50-mut (p.His1269Argfs2), indicating the frameshift mutation may markedly impair RAD50 protein expression or stability. Although recombinant human growth hormone significantly improved the patient's growth rate (from -3.35 SD to -1.28 SD), these variants may serve as a potential molecular basis for Nijmegen Breakage Syndrome-like disease and could also increase the risk of tumor formation. Treatment with recombinant human growth hormone was discontinued when the patient was 10 years and 3 months old. A five-year follow-up showed no evidence of a tumor was observed; The 15-year-old patient's height ceased to increase and remained at 151.5 cm. In the current study, we identified and characterized a patient with two RAD50 mutations. This report expands the clinical and genetic scope of RAD50 mutations. For the first time, it describes the response to unplanned recombinant human growth hormone therapy and the risk of long-term tumors. This report is intended to raise clinical awareness of the risk-benefit balance of recombinant human growth hormone therapy for syndromic short stature.
Expanding the mutational spectrum of RAD50: a case report of Nijmegen breakage syndrome-like disorder in a Chinese child.
Nijmegen breakage syndrome-like disorder (NBSLD) is a rare chromosomal instability syndrome caused by biallelic pathogenic variants in RAD50, which is a key component of the MRE11-RAD50-NBS1 (MRN) complex involved in DNA double-strand break repair. Merely few cases have been reported worldwide, and its phenotypic spectrum remains incompletely defined. We report a 6-year-old Chinese boy, who presented with bilateral cryptorchidism, severe microcephaly, growth retardation, multiple café-au-lait macules, brachydactyly, and distinctive craniofacial features, including a sloping forehead, midface prominence, and receding mandible. Mild intellectual impairment was confirmed on formal neurocognitive testing. The immunological assessment revealed borderline lymphopenia without overt immunodeficiency. Whole-exome sequencing (WES) identified compound heterozygosity for two variants in RAD50 (NM_005732.3): a paternally inherited frameshift variant c.2165_2166insT (p.Lys722Asnfs*6) and a maternally inherited splice-site variant c.3752 + 4_3752 + 7dup. The minigene splicing assay revealed that the latter disrupts normal splicing, leading to partial intron retention and a premature stop codon (p.Ile1252*). Based on the clinical features and molecular confirmation, the patient was diagnosed with NBSLD. Allogeneic hematopoietic stem cell transplantation (HSCT) has been proposed as a potential therapeutic option for DNA damage repair disorders. However, it is not presently required for this patient, who is managed with regular surveillance. The present case expands the clinical and mutational spectrum of RAD50-associated NBSLD. It emphasizes the importance of combining clinical assessment, genomic analysis, and functional assays for the accurate diagnosis of rare chromosomal breakage disorders.
[Research Progress in the Roles of MRE11-RAD50-NBS1 Complex and Human Diseases].
DNA is susceptible to various factors in vitro and in vivo and experience different forms of damage,among which double-strand break(DSB)is a deleterious form.To maintain the stability of genetic information,organisms have developed multiple mechanisms to repair DNA damage.Among these mechanisms,homologous recombination(HR)is praised for the high accuracy.The MRE11-RAD50-NBS1(MRN)complex plays an important role in HR and is conserved across different species.The knowledge on the MRN complex mainly came from the previous studies in Saccharomyces cerevisiae and Caenorhabditis elegans,while studies in the last decades have revealed the role of mammalian MRN complex in DNA repair of higher animals.In this review,we first introduces the MRN complex regarding the composition,structure,and roles in HR.In addition,we discuss the human diseases such as ataxia-telangiectasia-like disorder,Nijmegen breakage syndrome,and Nijmegen breakage syndrome-like disorder that are caused by dysfunctions in the MRN complex.Furthermore,we summarize the mouse models established to study the clinical phenotypes of the above diseases. 生物体的DNA常遭受着来自体外和体内各种因素的攻击,其中DNA双链断裂(DSB)是严重的一种DNA损伤方式。为了保证遗传信息的稳定性,生物体自身存在应对DNA损伤的修复机制。同源重组修复是精确的修复DSB的方式,MRE11-RAD50-NBS1(MRN)复合物是参与同源重组修复的关键蛋白,在不同物种之间存在保守性。从前关于MRN复合物的功能研究主要来源于酿酒酵母和线虫等低等生物,近些年来对哺乳动物MRN复合物的研究提示MRN复合物在高等动物DNA损伤修复中存在功能。本文综述了MRN复合物的组成和结构及其在DNA损伤同源重组修复中的功能,同时也介绍了MRN复合物异常所带来的人类疾病共济失调性毛细血管扩张综合征类似病症、奈梅亨断裂综合征和奈梅亨断裂综合征类似病症,并对这 3类DNA损伤修复缺陷疾病的临床表型和相关小鼠模型研究进行了总结。.
Bone Marrow Failure and Immunodeficiency Associated with Human RAD50 Variants.
The MRE11-RAD50-NBN (MRN) complex plays a key role in recognizing and signaling DNA double-strand breaks. Pathogenic variants in NBN and MRE11 give rise to the autosomal-recessive diseases, Nijmegen breakage syndrome (NBS) and ataxia telangiectasia-like disorder, respectively. The clinical consequences of pathogenic variants in RAD50 are incompletely understood. We aimed to characterize a newly identified RAD50 deficiency/NBS-like disorder (NBSLD) patient with bone marrow failure and immunodeficiency. We report on a girl with microcephaly, mental retardation, bird-like face, short stature, bone marrow failure and B-cell immunodeficiency. We searched for candidate gene by whole-exome sequencing and analyzed the cellular phenotype of patient-derived fibroblasts using immunoblotting, radiation sensitivity assays and lentiviral complementation experiments. Compound heterozygosity for two variants in the RAD50 gene (p.Arg83His and p.Glu485Ter) was identified in this patient. The expression of RAD50 protein and MRN complex formation was maintained in the cells derived from this patient. DNA damage-induced activation of the ATM kinase was markedly decreased, which was restored by the expression of wild-type (WT) RAD50. Radiosensitivity appeared inconspicuous in the patient-derived cell line as assessed by colony formation assay. The RAD50R83H missense substitution did not rescue the mitotic defect in complementation experiments using RAD50-deficient fibroblasts, whereas RAD50WT did. The RAD50E485X nonsense variant was associated with in-frame skipping of exon 10 (p.Glu485_545del). These findings indicate important roles of RAD50 in human bone marrow and immune cells. RAD50 deficiency/NBSLD can manifest as a distinct inborn error of immunity characterized by bone marrow failure and B-cell immunodeficiency.
Human RAD50 deficiency: Confirmation of a distinctive phenotype.
DNA double-strand breaks (DSBs) are highly toxic DNA lesions that can lead to chromosomal instability, loss of genes and cancer. The MRE11/RAD50/NBN (MRN) complex is keystone involved in signaling processes inducing the repair of DSB by, for example, in activating pathways leading to homologous recombination repair and nonhomologous end joining. Additionally, the MRN complex also plays an important role in the maintenance of telomeres and can act as a stabilizer at replication forks. Mutations in NBN and MRE11 are associated with Nijmegen breakage syndrome (NBS) and ataxia telangiectasia (AT)-like disorder, respectively. So far, only one single patient with biallelic loss of function variants in RAD50 has been reported presenting with features classified as NBS-like disorder. Here, we report a long-term follow-up of an unrelated patient with facial dysmorphisms, microcephaly, skeletal features, and short stature who is homozygous for a novel variant in RAD50. We could show that this variant, c.2524G > A in exon 15 of the RAD50 gene, induces aberrant splicing of RAD50 mRNA mainly leading to premature protein truncation and thereby, most likely, to loss of RAD50 function. Using patient-derived primary fibroblasts, we could show abnormal radioresistant DNA synthesis confirming pathogenicity of the identified variant. Immunoblotting experiments showed strongly reduced protein levels of RAD50 in the patient-derived fibroblasts and provided evidence for a markedly reduced radiation-induced AT-mutated signaling. Comparison with the previously reported case and with patients presenting with NBS confirms that RAD50 mutations lead to a similar, but distinctive phenotype.
Publicações recentes
Novel RAD50 variants lead to Nijmegen Breakage Syndrome-like disorder and unplanned recombinant human growth hormone treatment response.
Expanding the mutational spectrum of RAD50: a case report of Nijmegen breakage syndrome-like disorder in a Chinese child.
[Research Progress in the Roles of MRE11-RAD50-NBS1 Complex and Human Diseases].
Human RAD50 deficiency: Confirmation of a distinctive phenotype.
Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder.
📚 EuropePMC3 artigos no totalmostrando 5
Novel RAD50 variants lead to Nijmegen Breakage Syndrome-like disorder and unplanned recombinant human growth hormone treatment response.
Frontiers in endocrinologyExpanding the mutational spectrum of RAD50: a case report of Nijmegen breakage syndrome-like disorder in a Chinese child.
Gene[Research Progress in the Roles of MRE11-RAD50-NBS1 Complex and Human Diseases].
Zhongguo yi xue ke xue yuan xue bao. Acta Academiae Medicinae SinicaeBone Marrow Failure and Immunodeficiency Associated with Human RAD50 Variants.
Journal of clinical immunologyHuman RAD50 deficiency: Confirmation of a distinctive phenotype.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença síndrome de quebras de Nijmegen-like.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença síndrome de quebras de Nijmegen-like
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Novel RAD50 variants lead to Nijmegen Breakage Syndrome-like disorder and unplanned recombinant human growth hormone treatment response.
- Expanding the mutational spectrum of RAD50: a case report of Nijmegen breakage syndrome-like disorder in a Chinese child.
- [Research Progress in the Roles of MRE11-RAD50-NBS1 Complex and Human Diseases].Zhongguo yi xue ke xue yuan xue bao. Acta Academiae Medicinae Sinicae· 2024· PMID 38686720mais citado
- Bone Marrow Failure and Immunodeficiency Associated with Human RAD50 Variants.
- Human RAD50 deficiency: Confirmation of a distinctive phenotype.
- Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:240760(Orphanet)
- OMIM OMIM:613078(OMIM)
- MONDO:0013118(MONDO)
- GARD:17184(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55395613(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Doença síndrome de quebras de Nijmegen-like
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata