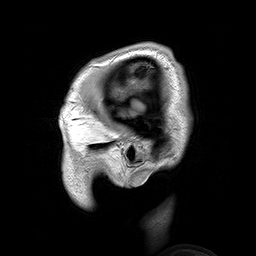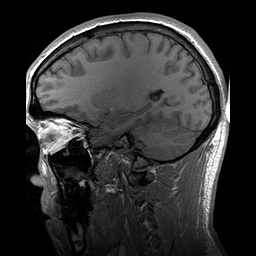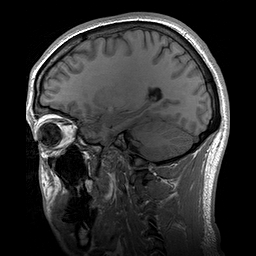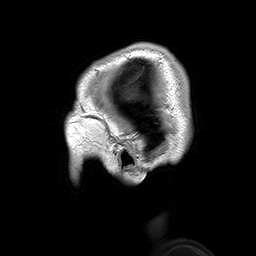
Hydrolethalus (HLS) é uma síndrome de malformação fetal grave caracterizada por características dismórficas craniofaciais, sistema nervoso central, coração, trato respiratório e anormalidades nos membros.
Introdução
O que você precisa saber de cara
Hydrolethalus (HLS) é uma síndrome de malformação fetal grave caracterizada por características dismórficas craniofaciais, sistema nervoso central, coração, trato respiratório e anormalidades nos membros.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 28 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 61 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Plays a role in ciliogenesis
CytoplasmCell projection, ciliumCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Hydrolethalus syndrome 1
A lethal syndrome characterized by polydactyly, central nervous system malformation, and hydrocephalus. The polydactyly is postaxial in the hands and preaxial in the feet. A highly characteristic hallux duplex is seen in almost no other situation. In half of the cases, a large atrioventricular communis defect of the heart is found. The pregnancy is characterized by hydramnios, which is often massive, and by preterm delivery.
Essential for hedgehog signaling regulation: acts both as a negative and positive regulator of sonic hedgehog (Shh) and Indian hedgehog (Ihh) pathways, acting downstream of SMO, through both SUFU-dependent and -independent mechanisms (PubMed:21633164). Involved in the regulation of microtubular dynamics. Required for proper organization of the ciliary tip and control of ciliary localization of SUFU-GLI2 complexes (By similarity). Required for localization of GLI3 to cilia in response to Shh. Neg
Cell projection, ciliumCytoplasm, cytoskeleton, cilium basal body
Variantes genéticas (ClinVar)
386 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 373 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hidroletal
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Centriole structural integrity defects are a crucial feature of hydrolethalus syndrome.
Hydrolethalus syndrome (HLS) is a lethal, autosomal recessive ciliopathy caused by the mutation of the conserved centriole protein HYLS1. How HYLS1 controls centriole function is poorly understood. Here, we show that mice harboring the HYLS1 disease mutation die shortly after birth and exhibit developmental defects that recapitulate several manifestations of HLS. These phenotypes arise from a loss of centriole integrity that causes tissue-specific defects in cilia assembly and function. We show that HYLS1 is recruited to the centriole by CEP120 and stabilizes the localization of centriole inner scaffold proteins that ensure the integrity of the centriolar microtubule wall. The HLS disease mutation reduced the centriole localization of HYLS1 and caused degeneration of the centriole distal end. We propose that tissue-specific defects in centriole integrity caused by the HYLS1 mutation prevent ciliogenesis and contribute to HLS phenotypes.
Hydrolethalus Syndrome: A Case of a Rare Congenital Disorder.
This is a fatal case of multiple complicated congenital anomalies displaying several symptoms consistent with hydrolethalus syndrome. The newborn's phenotype is characterized by a combination of serious anatomical abnormalities such as open-book cerebral hemispheres, defective lobulation of the lungs (one lobe on the left, two on the right), a smaller right kidney, a smooth cerebral surface, and a specific keyhole-shaped defect in the skull base, primarily associated with hydrocephalus.
Expanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome.
KIAA0586 variants have been associated with a wide range of ciliopathies, mainly Joubert syndrome (JS, OMIM #616490) and short-rib thoracic dysplasia syndrome (SRTD, OMIM #616546). However, the hypothesis that this gene is involved with hydrolethalus syndrome (HSL, OMIM #614120) and orofaciodigital syndrome IV (OMIM #258860) has already been raised. Ciliopathies' clinical features are often overlapped despite differing in phenotype severity. Besides KIAA0586, HYLS1 and KIF7 are also known for being causative of ciliopathies, indicating that all three genes may have similar or converging genomic pathways. Overall, the genotypic and phenotypic spectrum of ciliopathies becomes wider and conflicting while more and more new variants are added to this group of disorders' molecular pot. In this case report we discuss the first Brazilian individual clinically diagnosed with hydrolethalus syndrome and molecular findings that demonstrate the role of KIAA0586 as a causative gene of a group of genetic disorders. Also, recent reports on individuals with intronic and exonic variants combined leading to ciliopathies support our patient's molecular diagnosis. At the same time, we discuss variable expressivity and overlapping features in ciliopathies.
Novel HYLS1 variants associated with Joubert syndrome suggest potential genotype-phenotype correlates.
Joubert syndrome (JS) is an inherited neurodevelopmental ciliopathy with wide clinical and genetic heterogeneity, whose paradigmatic sign is a peculiar cerebellar and brainstem malformation known as the 'molar tooth sign'. Recessive pathogenic variants in the HYLS1 gene are associated with hydrolethalus syndrome (HLS), a severe disorder characterised by multiple developmental defects leading to intrauterine or perinatal death. However, HYLS1 biallelic variants were also reported in three individuals with JS.Here, we report a fourth patient with a purely neurological JS carrying two compound heterozygous missense variants in the HYLS1 gene. Notably, while all patients with lethal HLS had both variants falling within the highly conserved HYLS-1 Box, the four patients with milder JS phenotype featured at least one variant external to this evolutionary conserved domain, suggesting a possible correlation between the mutation site and the severity of the phenotype.
Retinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
Mutations in the KIF7 gene have been implicated in autosomal recessive conditions such as Joubert syndrome, acrocallosal syndrome, and fetal hydrolethalus, as well as in retinal degeneration and other ocular manifestations due to their effect on primary cilia. In this study, we report that the full-field electroretinogram (ERG) test showed non-recordable scotopic ERG responses, while photopic ERG responses were diminished bilaterally. This is a case report of a 62-year-old female patient with painless, progressive vision loss in both eyes. Fundus examination revealed a pale optic nerve head, vessel attenuation, and macular thinning without peripheral pigmentary changes. The full-field electroretinogram (ERG) test showed non-recordable scotopic ERG responses, while photopic ERG responses were diminished bilaterally. Based on these ocular findings, the patient was clinically diagnosed with retinitis pigmentosa (RP) sine pigmento. Genetic testing identified a pathogenic heterozygous mutation in the KIF7 gene with the variant c.61C>T (p.Arg21*). Our case suggests that this pathologic variant may be associated with RP sine pigmento. Further studies are warranted to better understand the role of the KIF7 gene in retinal dystrophies.
Publicações recentes
Centriole structural integrity defects are a crucial feature of hydrolethalus syndrome.
Hydrolethalus Syndrome: A Case of a Rare Congenital Disorder.
Novel HYLS1 variants associated with Joubert syndrome suggest potential genotype-phenotype correlates.
Expanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome.
Retinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
📚 EuropePMC50 artigos no totalmostrando 19
Centriole structural integrity defects are a crucial feature of hydrolethalus syndrome.
The Journal of cell biologyHydrolethalus Syndrome: A Case of a Rare Congenital Disorder.
Diagnostics (Basel, Switzerland)Novel HYLS1 variants associated with Joubert syndrome suggest potential genotype-phenotype correlates.
Journal of medical geneticsExpanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome.
International journal of molecular sciencesRetinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
CureusExercise Alters FBF1-Regulated Novel-miRNA-1135 Associated with Hydrolethalus Syndrome 1 in Rheumatoid Arthritis: A Preliminary Study.
MicroRNA (Shariqah, United Arab Emirates)In the Shadows of Rarity: A Case Report of Syndromic Cleft Lip and Palate!
CureusClinical and genetic spectrum from a prototype of ciliopathy: Joubert syndrome.
Clinical neurology and neurosurgeryThe first two non-Finnish HYLS1 variants: Expanding the phenotypic spectrum of hydrolethalus syndrome.
Clinical geneticsCiliopathy protein HYLS1 coordinates the biogenesis and signaling of primary cilia by activating the ciliary lipid kinase PIPKIγ.
Science advancesFunctional Analysis of Hydrolethalus Syndrome Protein HYLS1 in Ciliogenesis and Spermatogenesis in Drosophila.
Frontiers in cell and developmental biologyCentrioles initiate cilia assembly but are dispensable for maturation and maintenance in C. elegans.
The Journal of cell biologyThe hydrolethalus syndrome protein HYLS-1 regulates formation of the ciliary gate.
Nature communicationsA novel ICK mutation causes ciliary disruption and lethal endocrine-cerebro-osteodysplasia syndrome.
CiliaA novel HYLS1 homozygous mutation in living siblings with Joubert syndrome.
Clinical geneticsNovel KIF7 Mutation in a Tunisian Boy with Acrocallosal Syndrome: Case Report and Review of the Literature.
Molecular syndromology[Polydactyly, holoprosencephaly, cleft lip and cleft palate are not always what they seem: Case report].
Archivos argentinos de pediatriaNovel KIF7 missense substitutions in two patients presenting with multiple malformations and features of acrocallosal syndrome.
American journal of medical genetics. Part AMutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hidroletal.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hidroletal
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Centriole structural integrity defects are a crucial feature of hydrolethalus syndrome.
- Hydrolethalus Syndrome: A Case of a Rare Congenital Disorder.
- Expanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome.
- Novel HYLS1 variants associated with Joubert syndrome suggest potential genotype-phenotype correlates.
- Retinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2189(Orphanet)
- MONDO:0006037(MONDO)
- GARD:6683(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q5955105(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Hidroletal
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata