A hipoplasia pontocerebelar tipo 3 (PCH3), também conhecida como atrofia cerebelar com microcefalia progressiva (CLAM) é uma forma rara de hipoplasia pontocerebelar com transmissão autossômica recessiva caracterizada no período neonatal por hipotonia e dificuldade de deglutição e desde a infância por convulsões, atrofia óptica e baixa estatura, mas nenhum dos achados clínicos são específicos para PCH3.
Introdução
O que você precisa saber de cara
A hipoplasia pontocerebelar tipo 3 (PCH3), também conhecida como atrofia cerebelar com microcefalia progressiva (CLAM) é uma forma rara de hipoplasia pontocerebelar com transmissão autossômica recessiva caracterizada no período neonatal por hipotonia e dificuldade de deglutição e desde a infância por convulsões, atrofia óptica e baixa estatura, mas nenhum dos achados clínicos são específicos para PCH3.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 12 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 36 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisScaffold protein of the presynaptic cytomatrix at the active zone (CAZ) which is the place in the synapse where neurotransmitter is released (By similarity). After synthesis, participates in the formation of Golgi-derived membranous organelles termed Piccolo-Bassoon transport vesicles (PTVs) that are transported along axons to sites of nascent synaptic contacts (By similarity). At the presynaptic active zone, regulates the spatial organization of synaptic vesicle cluster, the protein complexes t
Presynaptic active zone
Pontocerebellar hypoplasia 3
A form of pontocerebellar hypoplasia, a disorder characterized by structural defects of the pons and cerebellum. Brain MRI shows an abnormally small cerebellum and brainstem, decreased cerebral white matter, and a thin corpus callosum. PCH3 features include seizures, short stature, optic atrophy, progressive microcephaly, severe developmental delay.
Variantes genéticas (ClinVar)
185 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 77 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipoplasia pontocerebelar tipo 3
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Pontocerebellar Hypoplasia Type 3 With Two Novel PCLO Gene Mutations: A Case Report.
Pontocerebellar hypoplasia Type III (PCH3) is a rare, autosomal recessive neurodegenerative disorder linked to mutations in the PCLO gene, previously reported only in Omani populations. It presents with progressive microcephaly, intractable epilepsy, optic atrophy, and severe developmental delay. Here, we report the first documented case of PCH3 in an 8-year-old Thai girl with two novel PCLO truncation mutations. The patient presented with intractable epilepsy from 2 months of age and severe global developmental delay. Whole exome sequencing identified compound heterozygous mutations in the PCLO gene: c.9018_9037del (p.Tyr3007Ter) and c.8456del (p.Ala2819GlufsTer2), both of which were inherited from heterozygous parents. These mutations are predicted to result in a loss of Piccolo protein function. This case expands the mutational spectrum of PCLO-related PCH3 and highlights the importance of advanced molecular diagnostics in understanding and managing this rare neurodegenerative disorder. Given the lack of curative therapies, early genetic diagnosis is crucial in guiding patient care and genetic counseling.
Synaptic logistics: The presynaptic scaffold protein Piccolo a nodal point tuning synaptic vesicle recycling, maintenance and integrity.
Properly working synapses are one important guarantor for a functional and healthy brain. They are small, densely packed structures, where information is transmitted through the release of neurotransmitters from synaptic vesicles (SVs). The latter cycle within the presynaptic terminal as they first fuse with the plasma membrane to deliver their neurotransmitter, and afterwards become recycled and prepared for a new release event. The synapse is an autonomous structure functioning mostly independent of the neuronal soma. Dysfunction in synaptic processes associated with local insults or genetic abnormalities can directly compromise synapse function and integrity and subsequently lead to the onset of neurodegenerative diseases. Therefore, measures need to be in place counteracting these threats for instance through the continuous replacement of old and damaged SV proteins. Interestingly recent studies show that the presynaptic scaffolding protein Piccolo contributes to health, function and integrity of synapses, as it mediates the delivery of synaptic proteins from the trans-Golgi network (TGN) towards synapses, as well as the local recycling and turnover of SV proteins within synaptic terminals. It can fulfill these various tasks through its multi-domain structure and ability to interact with numerous binding partners. In addition, Piccolo has recently been linked with the early onset neurodegenerative disease Pontocerebellar Hypoplasia Type 3 (PCH3) further underlying its importance for neuronal health. In this review, we will focus on Piccolo's contributions to synapse function, health and integrity and make a connection how those may contribute to the disease pattern of PCH3.
Loss of Piccolo Function in Rats Induces Cerebellar Network Dysfunction and Pontocerebellar Hypoplasia Type 3-like Phenotypes.
Piccolo, a presynaptic active zone protein, is best known for its role in the regulated assembly and function of vertebrate synapses. Genetic studies suggest a further link to several psychiatric disorders as well as Pontocerebellar Hypoplasia type 3 (PCH3). We have characterized recently generated Piccolo KO (Pclogt/gt ) rats. Analysis of rats of both sexes revealed a dramatic reduction in brain size compared with WT (Pclowt/wt ) animals, attributed to a decrease in the size of the cerebral cortical, cerebellar, and pontine regions. Analysis of the cerebellum and brainstem revealed a reduced granule cell layer and a reduction in size of pontine nuclei. Moreover, the maturation of mossy fiber afferents from pontine neurons and the expression of the α6 GABAA receptor subunit at the mossy fiber-granule cell synapse are perturbed, as well as the innervation of Purkinje cells by cerebellar climbing fibers. Ultrastructural and functional studies revealed a reduced size of mossy fiber boutons, with fewer synaptic vesicles and altered synaptic transmission. These data imply that Piccolo is required for the normal development, maturation, and function of neuronal networks formed between the brainstem and cerebellum. Consistently, behavioral studies demonstrated that adult Pclogt/gt rats display impaired motor coordination, despite adequate performance in tasks that reflect muscle strength and locomotion. Together, these data suggest that loss of Piccolo function in patients with PCH3 could be involved in many of the observed anatomical and behavioral symptoms, and that the further analysis of these animals could provide fundamental mechanistic insights into this devastating disorder.SIGNIFICANCE STATEMENT Pontocerebellar Hypoplasia Type 3 is a devastating developmental disorder associated with severe developmental delay, progressive microcephaly with brachycephaly, optic atrophy, seizures, and hypertonia with hyperreflexia. Recent genetic studies have identified non-sense mutations in the coding region of the PCLO gene, suggesting a functional link between this disorder and the presynaptic active zone. Our analysis of Piccolo KO rats supports this hypothesis, formally demonstrating that anatomical and behavioral phenotypes seen in patients with Pontocerebellar Hypoplasia Type 3 are also exhibited by these Piccolo deficient animals.
Teaching Video NeuroImages: Palatal tremor associated with SPG7 variants.
Severe intellectual disability, absence of language, epilepsy, microcephaly and progressive cerebellar atrophy related to the recurrent de novo variant p.(P139L) of the CAMK2B gene: A case report and brief review.
The CAMK2B gene encodes the β-subunit of calcium/calmodulin-dependent protein kinase II (CAMK2), an enzyme that has crucial roles in synaptic plasticity, especially in hippocampal and cerebellar neurons. Heterozygous variants in CAMK2B cause a rare neurodevelopmental disorder, with 40% of the reported cases sharing the same variant: c.416C>T, p.(P139L). This case report describes a 22-year-old patient with this recurrent variant, who presents with severe intellectual disability, absence of language, hypotonia, microcephaly, dysmorphic features, epilepsy, behavioral abnormalities, motor stereotypies, optic atrophy, and progressive cerebellar atrophy. Notably, this patient is the oldest reported so far and allows us to better delineate the clinical phenotype associated with this variant, adding clinical aspects never described before, such as epilepsy, optic atrophy, scoliosis, and neuroradiological changes characterized by progressive cerebellar atrophy.
Publicações recentes
Pontocerebellar Hypoplasia Type 3 With Two Novel PCLO Gene Mutations: A Case Report.
Synaptic logistics: The presynaptic scaffold protein Piccolo a nodal point tuning synaptic vesicle recycling, maintenance and integrity.
Loss of Piccolo Function in Rats Induces Cerebellar Network Dysfunction and Pontocerebellar Hypoplasia Type 3-like Phenotypes.
Pontocerebellar Hypoplasia Maps to Chromosome 7q11.23: An Autopsy Case Report of a Novel Genetic Variant.
Critical role for Piccolo in synaptic vesicle retrieval.
📚 EuropePMC251 artigos no totalmostrando 9
Pontocerebellar Hypoplasia Type 3 With Two Novel PCLO Gene Mutations: A Case Report.
Case reports in pediatricsSynaptic logistics: The presynaptic scaffold protein Piccolo a nodal point tuning synaptic vesicle recycling, maintenance and integrity.
Molecular and cellular neurosciencesSevere intellectual disability, absence of language, epilepsy, microcephaly and progressive cerebellar atrophy related to the recurrent de novo variant p.(P139L) of the CAMK2B gene: A case report and brief review.
American journal of medical genetics. Part AA patient with novel MBOAT7 variant: The cerebellar atrophy is progressive and displays a peculiar neurometabolic profile.
American journal of medical genetics. Part ATeaching Video NeuroImages: Palatal tremor associated with SPG7 variants.
NeurologyLoss of Piccolo Function in Rats Induces Cerebellar Network Dysfunction and Pontocerebellar Hypoplasia Type 3-like Phenotypes.
The Journal of neuroscience : the official journal of the Society for NeurosciencePontocerebellar Hypoplasia Maps to Chromosome 7q11.23: An Autopsy Case Report of a Novel Genetic Variant.
Case reports in pediatricsCritical role for Piccolo in synaptic vesicle retrieval.
eLifeMilder progressive cerebellar atrophy caused by biallelic SEPSECS mutations.
Journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipoplasia pontocerebelar tipo 3.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipoplasia pontocerebelar tipo 3
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Pontocerebellar Hypoplasia Type 3 With Two Novel PCLO Gene Mutations: A Case Report.
- Synaptic logistics: The presynaptic scaffold protein Piccolo a nodal point tuning synaptic vesicle recycling, maintenance and integrity.
- Loss of Piccolo Function in Rats Induces Cerebellar Network Dysfunction and Pontocerebellar Hypoplasia Type 3-like Phenotypes.The Journal of neuroscience : the official journal of the Society for Neuroscience· 2020· PMID 32122952mais citado
- Teaching Video NeuroImages: Palatal tremor associated with SPG7 variants.
- Severe intellectual disability, absence of language, epilepsy, microcephaly and progressive cerebellar atrophy related to the recurrent de novo variant p.(P139L) of the CAMK2B gene: A case report and brief review.
- Pontocerebellar Hypoplasia Maps to Chromosome 7q11.23: An Autopsy Case Report of a Novel Genetic Variant.
- Critical role for Piccolo in synaptic vesicle retrieval.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:97249(Orphanet)
- OMIM OMIM:608027(OMIM)
- MONDO:0011948(MONDO)
- GARD:10708(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18966154(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
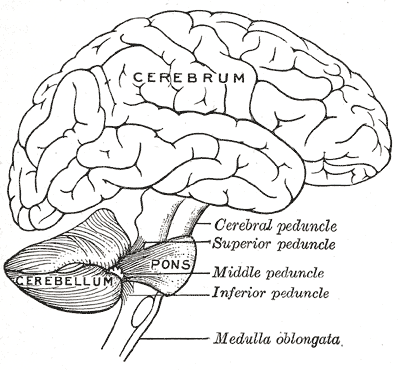
Hipoplasia pontocerebelar tipo 3
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata