Qualquer desenvolvimento incompleto da ponte e do cerebelo (partes do cérebro) que não faz parte de uma síndrome, e cuja causa é uma mutação no gene AMPD2.
Introdução
O que você precisa saber de cara
Qualquer desenvolvimento incompleto da ponte e do cerebelo (partes do cérebro) que não faz parte de uma síndrome, e cuja causa é uma mutação no gene AMPD2.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 14 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 36 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisAMP deaminase plays a critical role in energy metabolism. Catalyzes the deamination of AMP to IMP and plays an important role in the purine nucleotide cycle
Pontocerebellar hypoplasia 9
A form of pontocerebellar hypoplasia, a disorder characterized by structural defects of the pons and cerebellum, evident upon brain imaging. PCH9 features include severely delayed psychomotor development, progressive microcephaly, spasticity, seizures, and brain abnormalities, including brain atrophy, thin corpus callosum, and delayed myelination.
Variantes genéticas (ClinVar)
100 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 396 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipoplasia pontocerebelar tipo 9
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Pontocerebellar Hypoplasia Type 9: A Case Study Highlighting Distinctive Magnetic Resonance Imaging Features.
Pontocerebellar hypoplasia type 9 (PCH9) is a rare, autosomal, recessive, neurodevelopmental disorder caused by a mutation in the AMPD2 gene. Despite its rarity, it presents distinctive clinical and neuroradiological features. Diagnosing it is challenging yet crucial for appropriate management. We describe a 21-month-old boy with clinical and neuroradiological manifestations of the diagnosis, including characteristic signs such as an eight-configured midbrain and hypoplasia of the brainstem and cerebellar structures. Genetic evaluation confirmed homozygous missense mutations in the AMPD2 gene. This case highlights the pathognomonic neuroradiological features of pontocerebellar hypoplasia type 9 that point toward diagnosis.
Pontocerebellar Hypoplasia Type 9: A New Case with a Novel Mutation and Review of Literature.
Pontocerebellar hypoplasia type 9 (PCH-9) is a very rare autosomal recessive neurodegenerative disorder. Affected infants present early with severe developmental delay, spasticity, with the unique magnetic resonance imaging picture of thin corpus callosum, atrophied pons, and cerebellum. It is caused by loss of function mutations in the AMPD2 gene, encoding for the adenosine monophosphate deaminase enzyme-paralog 2. This gene is expressed in different somatic tissues with high level of expression in cerebellum and its encoded enzyme catalyzes a critical step in de novo biosynthesis of purines and its deficiency in the developing neurons severely affects neuronal differentiation and cell viability. We clinically evaluated an Emirati patient presented with severe developmental and growth delay, as well as corpus callosum agenesis and atrophy of brainstem and cerebellum. We performed exome sequencing, Sanger sequencing, and segregation analysis to identify the genetic cause of the phenotype, followed by in silico and in vitro analysis. We identified the novel variant (NM_004037.9:c.1471G > A) in AMPD2 gene leading to a single amino acid substitution (p.Gly491Arg) in adenosine monophosphate deaminase-2 enzyme. This variant is predicted to be pathogenic using several in silico tools, and resulted in a decrease in the enzyme function in the patient's polymorphonuclear cells by 82% (95% confidence interval: 73.3-91.7%, p = 0.029) compared with the control. This data establishes that the affected child is affected by PCH-9. Furthermore, we review all reported cases in literature to summarize the main clinical features of this rare disease.
Pontocerebellar hypoplasia type-9 due to a novel p.Arg503Ter truncating variant in AMPD2: a report from India.
Homozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
Pontocerebellar hypoplasia type 9 (PCH9) is an autosomal recessive neurodevelopmental disorder caused by pathogenic variants in the AMPD2 gene. We evaluated the son of a consanguineous couple who presented with profound hypotonia and global developmental delay. Other features included sensorineural hearing loss, asymmetric astigmatism, and high myopia. Clinical whole-exome sequence analysis identified a homozygous missense variant in AMPD2 (NM_001257360.1:c.2201C > T, p.[Pro734Leu]) that has not been previously reported. Given the strong phenotypic overlap with PCH9, including the identification of the typical "Figure 8" appearance of the brainstem on neuroimaging, we suspect this variant was causative of the neurodevelopmental disability in this individual. An additional homozygous nonsense variant in COL11A1 (NM_001854.4:c.1168G > T, p.[Glu390Ter]) was identified. Variants in this alternatively spliced region of COL11A1 have been identified to cause an autosomal recessive form of Stickler syndrome type 2 characterized by sensorineural hearing loss and eye abnormalities, but without musculoskeletal abnormalities. The COL11A1 variant likely also contributed to the individual's phenotype, suggesting two potentially relevant genetic findings. This challenging case highlights the importance of detailed phenotypic characterization when interpreting whole exome data.
CUGC for pontocerebellar hypoplasia type 9 and spastic paraplegia-63.
1. NAME OF DISEASE (SYNONYMS): Pontocerebellar hypoplasia type 9 (PCH9) and spastic paraplegia-63 (SPG63). 2. OMIM# OF THE DISEASE: 615809 and 615686. 3. NAME OF THE ANALYSED GENES OR DNA/CHROMOSOME SEGMENTS: AMPD2 at 1p13.3. 4. OMIM# OF THE GENE(S): 102771.
Publicações recentes
Pontocerebellar Hypoplasia Type 9: A Case Study Highlighting Distinctive Magnetic Resonance Imaging Features.
Pontocerebellar Hypoplasia Type 9: A New Case with a Novel Mutation and Review of Literature.
Pontocerebellar hypoplasia type-9 due to a novel p.Arg503Ter truncating variant in AMPD2: a report from India.
Neuroradiological findings in three cases of pontocerebellar hypoplasia type 9 due to AMPD2 mutation: typical MRI appearances and pearls for differential diagnosis.
Homozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
📚 EuropePMC251 artigos no totalmostrando 8
Pontocerebellar Hypoplasia Type 9: A Case Study Highlighting Distinctive Magnetic Resonance Imaging Features.
CureusPontocerebellar Hypoplasia Type 9: A New Case with a Novel Mutation and Review of Literature.
Journal of pediatric geneticsPontocerebellar hypoplasia type-9 due to a novel p.Arg503Ter truncating variant in AMPD2: a report from India.
Acta neurologica BelgicaNeuroradiological findings in three cases of pontocerebellar hypoplasia type 9 due to AMPD2 mutation: typical MRI appearances and pearls for differential diagnosis.
Quantitative imaging in medicine and surgeryHomozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
American journal of medical genetics. Part ACUGC for pontocerebellar hypoplasia type 9 and spastic paraplegia-63.
European journal of human genetics : EJHGClinical and genetic spectrum of AMPD2-related pontocerebellar hypoplasia type 9.
European journal of human genetics : EJHGA novel AMPD2 mutation outside the AMP deaminase domain causes pontocerebellar hypoplasia type 9.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipoplasia pontocerebelar tipo 9.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipoplasia pontocerebelar tipo 9
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Pontocerebellar Hypoplasia Type 9: A Case Study Highlighting Distinctive Magnetic Resonance Imaging Features.
- Pontocerebellar Hypoplasia Type 9: A New Case with a Novel Mutation and Review of Literature.
- Pontocerebellar hypoplasia type-9 due to a novel p.Arg503Ter truncating variant in AMPD2: a report from India.
- Homozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
- CUGC for pontocerebellar hypoplasia type 9 and spastic paraplegia-63.
- Neuroradiological findings in three cases of pontocerebellar hypoplasia type 9 due to AMPD2 mutation: typical MRI appearances and pearls for differential diagnosis.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:369920(Orphanet)
- OMIM OMIM:615809(OMIM)
- MONDO:0014351(MONDO)
- GARD:17590(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18966162(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
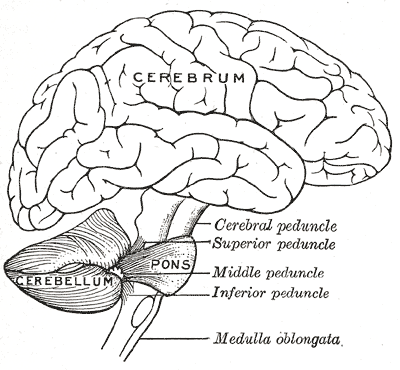
Hipoplasia pontocerebelar tipo 9
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata