A osteogênese imperfeita tipo III é um tipo grave de osteogênese imperfeita (OI), uma doença genética caracterizada por aumento da fragilidade óssea, baixa massa óssea e suscetibilidade a fraturas ósseas. Os principais sinais do tipo III incluem estatura muito baixa, face triangular, escoliose grave, esclera acinzentada e dentinogênese imperfeita (DI).
Introdução
O que você precisa saber de cara
A osteogênese imperfeita tipo III é um tipo grave de osteogênese imperfeita (OI), uma doença genética caracterizada por aumento da fragilidade óssea, baixa massa óssea e suscetibilidade a fraturas ósseas. Os principais sinais do tipo III incluem estatura muito baixa, face triangular, escoliose grave, esclera acinzentada e dentinogênese imperfeita (DI).
Tem tratamento?
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 10 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 27 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
13 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked recessive.
Curadoria gene-doença
fontes oficiaisPrecursor of the transcription factor form (Processed cyclic AMP-responsive element-binding protein 3-like protein 1), which is embedded in the endoplasmic reticulum membrane with N-terminal DNA-binding and transcription activation domains oriented toward the cytosolic face of the membrane (PubMed:12054625, PubMed:16417584, PubMed:25310401). In response to ER stress or DNA damage, transported to the Golgi, where it is cleaved in a site-specific manner by resident proteases S1P/MBTPS1 and S2P/MBT
Endoplasmic reticulum membraneNucleus
Osteogenesis imperfecta 16
An autosomal recessive form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI16 is a severe form.
Metalloprotease that plays key roles in regulating the formation of the extracellular matrix (ECM) via processing of various precursor proteins into mature functional enzymes or structural proteins (PubMed:33206546). Thereby participates in several developmental and physiological processes such as cartilage and bone formation, muscle growth and homeostasis, wound healing and tissue repair (PubMed:32636307, PubMed:33169406). Roles in ECM formation include cleavage of the C-terminal propeptides fr
Golgi apparatus, trans-Golgi networkSecreted, extracellular space, extracellular matrixSecreted
Osteogenesis imperfecta 13
An autosomal recessive form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI13 is characterized by normal teeth, faint blue sclerae, severe growth deficiency, severe bone deformity, and recurrent fractures affecting both upper and lower limbs.
Basement membrane-associated chondroitin sulfate proteoglycan (CSPG). Has prolyl 3-hydroxylase activity catalyzing the post-translational formation of 3-hydroxyproline in -Xaa-Pro-Gly- sequences in collagens, especially types IV and V. May be involved in the secretory pathway of cells. Has growth suppressive activity in fibroblasts
Endoplasmic reticulumSecreted, extracellular space, extracellular matrix
Osteogenesis imperfecta 8
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI8 is characterized by disproportionate short stature, shortening of the long bones, white sclerae, a round face and a short barrel-shaped chest.
Cytoplasmic non-canonical poly(A) RNA polymerase that catalyzes the transfer of one adenosine molecule from an ATP to an mRNA poly(A) tail bearing a 3'-OH terminal group and participates in the cytoplasmic polyadenylation (PubMed:33882302). Polyadenylates mRNA encoding extracellular matrix constituents and other genes crucial for bone mineralization and during osteoblast mineralization, mainly focuses on ER-targeted mRNAs (By similarity)
Cytoplasm
Osteogenesis imperfecta 18
An autosomal recessive form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI18 is a severe form characterized by congenital bowing of the lower limb, wormian bones, blue sclerae, vertebral collapses and multiple fractures in the first years of life.
PPIases accelerate the folding of proteins during protein synthesis
Endoplasmic reticulum lumen
Osteogenesis imperfecta 11
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI11 is an autosomal recessive form.
PPIase that catalyzes the cis-trans isomerization of proline imidic peptide bonds in oligopeptides and may therefore assist protein folding
VirionEndoplasmic reticulum lumenMelanosome
Osteogenesis imperfecta 9
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI9 is a severe autosomal recessive form of the disorder.
Necessary for efficient 3-hydroxylation of fibrillar collagen prolyl residues
Secreted, extracellular space, extracellular matrix
Osteogenesis imperfecta 7
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI7 is an autosomal recessive, severe form. Multiple fractures are present at birth and patients have short stature, short humeri and femora, coxa vara, and white sclera. Dentinogenesis imperfecta is absent. Death can occur in the perinatal period due to secondary respiratory insufficiency.
Ligand for members of the frizzled family of seven transmembrane receptors (Probable). Acts in the canonical Wnt signaling pathway by promoting beta-catenin-dependent transcriptional activation (PubMed:23499309, PubMed:23656646, PubMed:26902720, PubMed:28528193). In some developmental processes, is also a ligand for the coreceptor RYK, thus triggering Wnt signaling (By similarity). Plays an essential role in the development of the embryonic brain and central nervous system (CNS) (By similarity).
Secreted, extracellular space, extracellular matrixSecreted
Osteoporosis
A systemic skeletal disorder characterized by decreased bone mass and deterioration of bone microarchitecture without alteration in the composition of bone. The result is fragile bones and an increased risk of fractures, even after minimal trauma. Osteoporosis is a chronic condition of multifactorial etiology and is usually clinically silent until a fracture occurs.
Zinc metalloprotease that mediates intramembrane proteolysis of proteins such as ATF6, ATF6B, SREBF1/SREBP1 and SREBF2/SREBP2 (PubMed:10805775, PubMed:11163209). Catalyzes the second step in the proteolytic activation of the sterol regulatory element-binding proteins (SREBPs) SREBF1/SREBP1 and SREBF2/SREBP2: cleaves SREBPs within the first transmembrane segment, thereby releasing the N-terminal segment with a portion of the transmembrane segment attached (PubMed:10805775, PubMed:27380894, PubMed
MembraneCytoplasmGolgi apparatus membrane
IFAP syndrome 1, with or without Bresheck syndrome
An X-linked syndrome characterized by a peculiar triad of follicular ichthyosis, total or subtotal atrichia, and photophobia of varying degree. Histopathologically, the epidermal granular layer is generally well-preserved or thickened at the infundibulum. Hair follicles are poorly developed and tend to be surrounded by an inflammatory infiltrate. A subgroup of patients is described with lamellar rather than follicular ichthyosis. Non-consistent features may include growth and psychomotor retardation, aganglionic megacolon, seizures and nail dystrophy.
Neurotrophic protein; induces extensive neuronal differentiation in retinoblastoma cells. Potent inhibitor of angiogenesis. As it does not undergo the S (stressed) to R (relaxed) conformational transition characteristic of active serpins, it exhibits no serine protease inhibitory activity
SecretedMelanosome
Osteogenesis imperfecta 6
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI6 is a severe, autosomal recessive form compatible with OI type III in the Sillence classification.
Type I collagen is a member of group I collagen (fibrillar forming collagen)
Secreted, extracellular space, extracellular matrix
Ehlers-Danlos syndrome, arthrochalasia type, 2
A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSARTH2 is an autosomal dominant condition characterized by frequent congenital hip dislocation and extreme joint laxity with recurrent joint subluxations and minimal skin involvement.
Binds specifically to collagen. Could be involved as a chaperone in the biosynthetic pathway of collagen
Endoplasmic reticulum lumen
Osteogenesis imperfecta 10
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI10 is an autosomal recessive form characterized by multiple bone deformities and fractures, generalized osteopenia, dentinogenesis imperfecta, and blue sclerae.
Type I collagen is a member of group I collagen (fibrillar forming collagen)
Secreted, extracellular space, extracellular matrix
Caffey disease
An autosomal dominant disorder characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age.
Medicamentos e terapias
Mecanismo: Vitamin D receptor agonist
Mecanismo: Sclerostin inhibitor
Mecanismo: Farnesyl diphosphate synthase inhibitor
Mecanismo: Farnesyl diphosphate synthase inhibitor
Variantes genéticas (ClinVar)
2.810 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 7.473 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
43 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Osteogenesis imperfecta tipo 3
Centros de Referência SUS
24 centros habilitados pelo SUS para Osteogenesis imperfecta tipo 3
Centros para Osteogenesis imperfecta tipo 3
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
3 ensaios clínicos encontrados, 1 ativos.
Publicações mais relevantes
Co-Occurrence of Osteogenesis Imperfecta Type III and Chronic Abruption-Oligohydramnios Sequence: A Case Report Suggesting a Possible Role of Type I Collagen Fragility.
Pregnancy complicated by severe osteogenesis imperfecta (OI) is rare, and chronic abruption-oligohydramnios sequence (CAOS) is an uncommon obstetric disorder characterized by persistent bleeding and oligohydramnios without membrane rupture. To our knowledge, this is the first report describing the coexistence of type III OI and CAOS. A 24-year-old woman with OI developed recurrent bleeding early in pregnancy. Genetic testing revealed a novel COL1A1 splice-site variant (c.1876-2A>C). Ultrasonography showed a circumferential subchorionic hematoma and progressive fluid loss, leading to CAOS diagnosis at 19 weeks. Despite amnioinfusion, the amniotic cavity collapsed, and pregnancy was terminated via cesarean section at 20 weeks. Placental pathology showed hemosiderin and fibrin deposition. In this case, both functional and structural factors possibly associated with type I collagen abnormalities may have contributed to CAOS development. Although causality cannot be proven, persistent bleeding in severe OI may warrant greater caution for progression to CAOS.
Magnetically Controlled Intramedullary Compression Nailing for Femoral Nonunion in Osteogenesis Imperfecta: A Case Report.
A 51-year-old nonambulatory woman with type III osteogenesis imperfecta sustained a diaphyseal femur fracture during a transfer, distal to a childhood-placed intramedullary pin. At the time of presentation, the fracture was a painful atrophic nonunion, preventing effective transfer. Retrograde nailing with a motorized magnetic lengthening nail, inserted with preset distraction, allowed gradual compression, solid union, and pain relief. This report describes the method of off-label lengthening nail modification used for gradual compression of a femoral nonunion site, achieving healing in pathologic bone.
A C-Propeptide Variant in COL1A1 Potentially Perturbing Disulfide Bonding in Osteogenesis Imperfecta Type III.
Osteogenesis imperfecta (OI) is a genetic disorder characterized by bone fragility and frequent fractures. The disorder is caused by pathogenic variants in genes that encode type I collagen. The COL1A1 gene encodes the pro-α1(I) collagen chain, which assembles with another pro-α1(I) chain and one pro-α2(I) chain to form a heterotrimeric triple helix. The C-terminal propeptide (C-propeptide) contributes to molecular recognition and initiation of triple helix formation, thereby promoting correct chain alignment during procollagen biosynthesis. Variants in this region may disrupt chain association and folding, and impair collagen assembly. Here, we present the case of a Japanese male infant who presented with respiratory distress, pulmonary hypoplasia, recurrent fractures, and hearing loss. Radiographic examination revealed slender long bones with multiple acute and healing fractures in the ribs, clavicles, and limbs, as well as Wormian bones. Genetic analysis identified a de novo heterozygous COL1A1 variant, NM_000088.4:c.4223A>G, p.(Tyr1408Cys). Based on the clinical and molecular findings, the patient was diagnosed with OI type III. The patient received bisphosphonate therapy and respiratory support with continuous positive airway pressure. In silico structural modeling suggested that Tyr1408 is located near the C-propeptide interface, where it may contribute to local stability through aromatic and hydrogen-bonding interactions. Substitution by cysteine could alter the local folding environment and inter-chain interactions. Presently, the report provides clinical and structure-based context for how cysteine substitutions in the C-terminal region of COL1A1 may be associated with severe OI phenotypes.
Lung volumetry of osteogenesis imperfecta type 3 subjects is not correlated with thoracic scoliosis and anthropometric data.
This study aimed to investigate the relationship between lung volumetry, thoracic scoliosis, and anthropometric data (height, weight, BMI) in patients with Osteogenesis Imperfecta (OI) Type 3. Three hypotheses were tested: H1 predicted lower lung volumes in patients with OI Type 3 compared to controls, H2 predicted differences between right and left lung volumes in patients with OI Type 3 due to chest deformities, and H3 predicted a correlation between lung volumes in patients with OI Type 3 and their thoracic scoliosis and anthropometric data. Age, biological sex, weight, height, body mass index (BMI), Cobb angle of thoracic scoliosis, left and right lung volumes, and total lung volume were recorded. CT scans were performed on all participants, and lung volumetry was analysed using specialised software. The intraclass correlation coefficient was used to assess measurement reliability, and statistical analysis was conducted to examine correlations between variables. Patients with OI had significantly lower total lung volumes than controls (p < 0.001). However, no significant correlation was found between lung volumetry and scoliosis (r =- 0.406; p = 0.244), age (r = 0.201; p = 0.578), height (r = 0.479; p = 0.162), weight (r = 0.358; p = 0.310), or BMI (r = - 0.042; p = 0.907) in OI patients. In the control group, significant correlations were observed between lung volume and height (r = 0.756; p = 0.011) and weight (r = 0.638; p = 0.047). OI type 3 patients have lower lung volumes than healthy subjects, but have no left and right lung volume differences. In addition, they did not present any correlation between lung volumes and scoliosis, height, weight, and body mass index.
The impact of foot orthoses on gait in children with Osteogenesis Imperfecta type I, III and IV - a cross-sectional study.
For children with Osteogenesis Imperfecta (OI), a rare genetic bone disease, walking can be difficult to carry out due to a combination of bone fragility and deformity, muscle weakness, joint hypermobility, and pain. Bisphosphonate treatment has facilitated more children being able to walk, but for many, foot and ankle hypermobility is a limiting factor. Current evidence on foot orthoses in children with OI is sparse. This study aimed to evaluate gait characteristics in children with OI walking barefoot as compared to walking with foot orthoses. Twenty-three children with OI and hypermobility (mean age 8.3 ± 3.0 years) were included in this cross-sectional study. Children conducted three-dimensional gait analysis barefoot, and with foot orthoses and appropriate foot wear (stable yet light-weight), respectively. Walking speed, step length, lower limb kinematics and kinetics were collected. Differences in gait characteristics between test conditions were evaluated using paired sample t-tests. When walking with foot orthoses, the external foot progression angle was reduced, peak ankle dorsiflexion angle increased, and peak plantarflexion moment increased as compared to barefoot. No difference was found in walking speed between test conditions, however, children with OI walked with longer steps with foot orthoses as compared to barefoot. The observed gait alterations suggest that foot orthoses, aiming to support the foot and ankle joint, contributed to reduced overall foot rotation as measured by external foot progression, increased peak plantarflexion moment, and increased step length. In a wider perspective, the ability to walk provides the opportunity to be physically active, and thereby increase skeletal loading and prevent fractures, thus, foot orthoses may be an important treatment option to consider in children with OI. III.
Publicações recentes
Lung volumetry of osteogenesis imperfecta type 3 subjects is not correlated with thoracic scoliosis and anthropometric data.
Multidisciplinary Management of Pregnancy in Patients With Osteogenesis Imperfecta Type 3.
Point of Care Ultrasound Identification of Multiple Rib Fractures in a Pediatric Patient with Osteogenesis Imperfecta Type 3.
Osteogenesis Imperfecta Type 3 in a 10-Year-Old Child With Acute Respiratory Distress Syndrome.
Hip Dysplasia and Osteogenesis Imperfecta: A Case Report.
📚 EuropePMC4.738 artigos no totalmostrando 47
Co-Occurrence of Osteogenesis Imperfecta Type III and Chronic Abruption-Oligohydramnios Sequence: A Case Report Suggesting a Possible Role of Type I Collagen Fragility.
The journal of obstetrics and gynaecology researchMagnetically Controlled Intramedullary Compression Nailing for Femoral Nonunion in Osteogenesis Imperfecta: A Case Report.
JBJS case connectorA C-Propeptide Variant in COL1A1 Potentially Perturbing Disulfide Bonding in Osteogenesis Imperfecta Type III.
Congenital anomaliesLung volumetry of osteogenesis imperfecta type 3 subjects is not correlated with thoracic scoliosis and anthropometric data.
Orphanet journal of rare diseasesThe impact of foot orthoses on gait in children with Osteogenesis Imperfecta type I, III and IV - a cross-sectional study.
BMC musculoskeletal disordersMultidisciplinary Management of Pregnancy in Patients With Osteogenesis Imperfecta Type 3.
The Permanente journalCross-sectional and longitudinal analysis of bone age maturation during peri-pubertal growth in children with type I, III and IV osteogenesis imperfecta.
BoneCraniofacial dysmorphism of osteogenesis imperfecta mouse and effect of cathepsin K knockout: Preliminary craniometry observations.
Morphologie : bulletin de l'Association des anatomistesMonochorionic twin pregnancy in a patient with type III osteogenesis imperfecta: a multidisciplinary challenge.
BMJ case reportsDental anomalies in individuals with osteogenesis imperfecta: a systematic review and meta-analysis of prevalence and comparative studies.
Journal of applied oral science : revista FOBRespiratory function of children and adolescents with osteogenesis imperfecta: respiratory muscle strength, forced vital capacity, and peak expiratory flow.
Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao PauloSkeletal outcomes of patients with osteogenesis imperfecta during drug holiday of bisphosphonates: a real-world study.
Frontiers in endocrinologyAnthropometrics of Polish children with osteogenesis imperfecta: a single-centre retrospective cohort study.
BMC pediatricsCraniofacial morphology in adults with osteogenesis imperfecta-A cross-sectional study.
Orthodontics & craniofacial researchFunctional status of individuals with osteogenesis imperfecta: data from a reference center.
Jornal de pediatriaPoint of Care Ultrasound Identification of Multiple Rib Fractures in a Pediatric Patient with Osteogenesis Imperfecta Type 3.
Children (Basel, Switzerland)Pubertal growth in osteogenesis imperfecta caused by pathogenic variants in COL1A1/COL1A2.
Genetics in medicine : official journal of the American College of Medical GeneticsPrevalence of scoliosis and impaired pulmonary function in patients with type III osteogenesis imperfecta.
European spine journal : official publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research SocietyOsteogenesis Imperfecta Type 3 in a 10-Year-Old Child With Acute Respiratory Distress Syndrome.
CureusHip Dysplasia and Osteogenesis Imperfecta: A Case Report.
JBJS case connectorPediatric Outcomes Data Collection Instrument is a Useful Patient-Reported Outcome Measure for Physical Function in Children with Osteogenesis Imperfecta.
Genetics in medicine : official journal of the American College of Medical GeneticsSurgical Treatment of Bilateral Tibia Deformity in a 9-Year-Old Child Suffering from Osteogenesis Imperfecta Type III: A Case Report.
The American journal of case reportsScoliosis and Cardiopulmonary Outcomes in Osteogenesis Imperfecta Patients.
SpineElastic intramedullary nailing of the femur fracture in patients affected by osteogenesis imperfecta type 3: Indications, limits and pitfalls.
InjuryPersonalized surgery approach in severe form of osteogenesis imperfecta type III: point of view.
Journal of pediatric orthopedics. Part BAnesthesia in children with osteogenesis imperfecta: Retrospective chart review of 83 patients and 205 anesthetics over 7 years.
Paediatric anaesthesiaLongitudinal growth curves for children with classical osteogenesis imperfecta (types III and IV) caused by structural pathogenic variants in type I collagen.
Genetics in medicine : official journal of the American College of Medical GeneticsIncidence and treatment of femur fractures in adults with osteogenesis imperfecta: an analysis of an expert clinic of 216 patients.
European journal of trauma and emergency surgery : official publication of the European Trauma SocietyTreatment of tibial deformities with the Fassier-Duval telescopic nail and minimally invasive percutaneous osteotomies in patients with osteogenesis imperfecta type III.
Journal of pediatric orthopedics. Part BMutations in the COL1A1 and COL1A2 genes associated with osteogenesis imperfecta (OI) types I or III.
Acta biochimica PolonicaIntraoperative bleeding in patients with osteogenesis imperfecta type III treated by Fassier-Duval femoral rodding: analysis of risk factors.
Journal of pediatric orthopedics. Part BExome sequencing reveals a novel homozygous splice site variant in the WNT1 gene underlying osteogenesis imperfecta type 3.
Pediatric researchOsteogenesis imperfecta type 3 in South Africa: Causative mutations in FKBP10.
South African medical journal = Suid-Afrikaanse tydskrif vir geneeskundeCombined Spinal-Epidural Anesthesia With Dexmedetomidine-Based Sedation for Multiple Corrective Osteotomies in a Child With Osteogenesis Imperfecta Type III: A Case Report.
A & A case reportsOsteogenesis imperfecta complicated with renal hypoplasia leads to chronic kidney disease.
Pediatrics international : official journal of the Japan Pediatric SocietySURGICAL MANAGEMENT OF RETINAL DETACHMENT IN OSTEOGENESIS IMPERFECTA: CASE REPORT AND REVIEW OF THE LITERATURE.
Retinal cases & brief reportsOsteogenesis imperfecta type III in South Africa: Psychosocial challenges.
South African medical journal = Suid-Afrikaanse tydskrif vir geneeskundePamidronate treatment for osteogenesis imperfecta in black South Africans.
South African medical journal = Suid-Afrikaanse tydskrif vir geneeskundeChildren with severe Osteogenesis imperfecta and short stature present on average with normal IGF-I and IGFBP-3 levels.
Journal of pediatric endocrinology & metabolism : JPEMCross-sectional and longitudinal growth patterns in osteogenesis imperfecta: implications for clinical care.
Pediatric researchGenetic drift: A case of abuse.
American journal of medical genetics. Part C, Seminars in medical geneticsSuccessful bone-anchored hearing aid implantation in a patient with osteogenesis imperfecta.
The Journal of laryngology and otologyOsteogenesis imperfecta type III and hypogonadotropic hypogonadism result in severe bone loss: a case report.
Wiener medizinische Wochenschrift (1946)Multidisciplinary Treatment of Severe Osteogenesis Imperfecta: Functional Outcomes at Skeletal Maturity.
Archives of physical medicine and rehabilitation[Late discovery of type III osteogenesis imperfecta: about a case].
The Pan African medical journalMutation Analysis of COL1A1 and COL1A2 in Fetuses with Osteogenesis Imperfecta Type II/III.
Gynecologic and obstetric investigation[Characteristics of anesthesia in patients with osteogenesis imperfecta undergoing orthopedic surgical procedures].
Lijecnicki vjesnikAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Osteogenesis imperfecta tipo 3.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Osteogenesis imperfecta tipo 3
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Co-Occurrence of Osteogenesis Imperfecta Type III and Chronic Abruption-Oligohydramnios Sequence: A Case Report Suggesting a Possible Role of Type I Collagen Fragility.
- Magnetically Controlled Intramedullary Compression Nailing for Femoral Nonunion in Osteogenesis Imperfecta: A Case Report.
- A C-Propeptide Variant in COL1A1 Potentially Perturbing Disulfide Bonding in Osteogenesis Imperfecta Type III.
- Lung volumetry of osteogenesis imperfecta type 3 subjects is not correlated with thoracic scoliosis and anthropometric data.
- The impact of foot orthoses on gait in children with Osteogenesis Imperfecta type I, III and IV - a cross-sectional study.
- Multidisciplinary Management of Pregnancy in Patients With Osteogenesis Imperfecta Type 3.
- Point of Care Ultrasound Identification of Multiple Rib Fractures in a Pediatric Patient with Osteogenesis Imperfecta Type 3.
- Osteogenesis Imperfecta Type 3 in a 10-Year-Old Child With Acute Respiratory Distress Syndrome.
- Hip Dysplasia and Osteogenesis Imperfecta: A Case Report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:216812(Orphanet)
- OMIM OMIM:259420(OMIM)
- MONDO:0009804(MONDO)
- GARD:8695(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q27674944(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
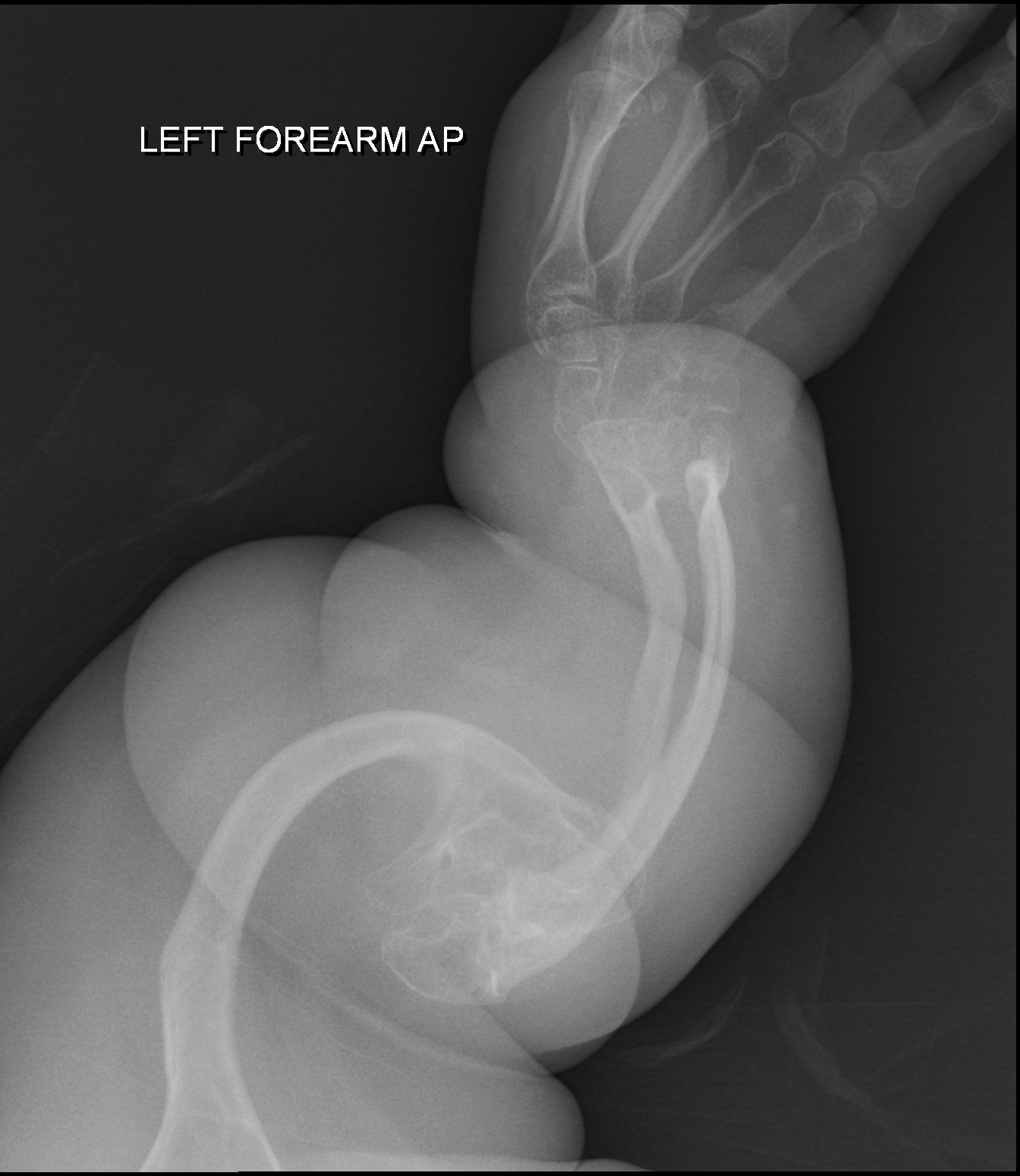
Osteogenesis imperfecta tipo 3
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet
- Ensaios clínicos
- fonte: ClinicalTrials.gov