Qualquer síndrome de Bartter em que a causa da doença seja uma mutação no gene KCNJ1.
Introdução
O que você precisa saber de cara
Qualquer síndrome de Bartter em que a causa da doença seja uma mutação no gene KCNJ1.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 25 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Curadoria gene-doença
fontes oficiaisInward rectifier potassium channels are characterized by a greater tendency to allow potassium to flow into the cell rather than out of it. Their voltage dependence is regulated by the concentration of extracellular potassium; as external potassium is raised, the voltage range of the channel opening shifts to more positive voltages. The inward rectification is mainly due to the blockage of outward current by internal magnesium. This channel is activated by internal ATP and can be blocked by exte
Cell membrane
Bartter syndrome 2, antenatal
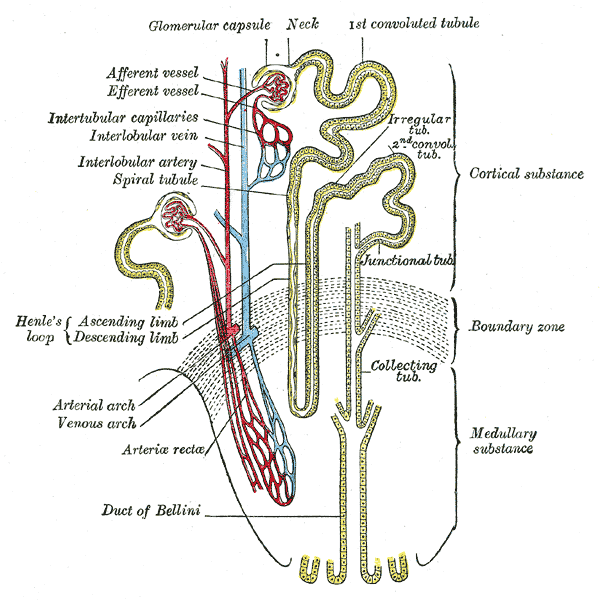
A form of Bartter syndrome, an autosomal recessive disorder characterized by impaired salt reabsorption in the thick ascending loop of Henle with pronounced salt wasting, hypokalemic metabolic alkalosis, and varying degrees of hypercalciuria. BARTS2 is a life-threatening condition beginning in utero, with marked fetal polyuria that leads to polyhydramnios and premature delivery. Another hallmark is a marked hypercalciuria and, as a secondary consequence, the development of nephrocalcinosis and osteopenia.
Variantes genéticas (ClinVar)
177 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 140 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Bartter tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Phenotypes and the Importance of Genetic Analysis in Adult Patients with Nephrolithiasis and/or Nephrocalcinosis: A Single-Center Experience.
Molecular analysis in patients with nephrolithiasis (NL) and/or nephrocalcinosis (NC) enables more accurate evaluation of underlying etiologies. The existing clinical evidence regarding genetic testing in adults with NL comprises only a few cohort studies. We retrospectively analyzed 49 adult patients diagnosed with NL and/or NC from a single center, on whom we performed a genetic test using a nephrolithiasis panel. We reviewed the phenotype of the patients and compared the cases with positive and negative molecular diagnosis. In total, 49 adult patients with NL and/or NC underwent genetic testing. Of the tested patients, 29 (59.2%) patients had 24 abnormal variants in 14 genes. Mendelian diseases were diagnosed in 14 (28.6%) cases: cystinuria (SLC3A1, SLC7A9; n = 4), hereditary distal renal tubular acidosis (SLC4A1; n = 3), Dent disease (CLCN5; n = 2), familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (CLDN16; n = 1), infantile hypercalcemia type 1 (CYP24A1; n = 1), primary hyperoxaluria type 1 (AGXT; n = 1), Bartter syndrome type 2 (KCNJ1; n = 1), and autosomal dominant tubulointerstitial kidney disease (UMOD; n = 1). Eight (16.3%) patients had pathogenic or likely pathogenic monoallelic variants as predisposing factors for NL and/or NC, and seven (14.3%) had biallelic or monoallelic variants of uncertain significance. Patients with positive genetic tests had a lower estimated glomerular filtration rate (p = 0.03) and more frequent NL associated with NC (p = 0.007) and were unlikely to have arterial hypertension (p = 0.03) when compared with patients with negative tests. Our study shows an increased effectiveness of molecular diagnosis and highlights the benefits of genetic testing. NL associated with NC and the presence of chronic kidney disease are the characteristics that should prompt the clinician to suspect an inherited form of NL and/or NC.
Late onset Bartter syndrome: Bartter syndrome type 2 presenting with isolated nephrocalcinosis and high parathyroid hormone levels mimicking primary hyperparathyroidism.
Nephrocalcinosis is associated with conditions that cause hypercalcemia and the increased urinary excretion of calcium, phosphate, and/or oxalate. A monogenic etiology is found in almost 30% of childhood-onset nephrocalcinosis which is also a common manifestation of primary hyperparathyroidism. We discuss a child with nephrocalcinosis and features mimicking primary hyperparathyroidism. A 7-year-old girl presented with nephrocalcinosis. Hypercalciuria, hyperphosphaturia, mild hypercalcemia, hypophosphatemia and elevated parathyroid hormone levels along with normal serum creatinine and absence of hypokalemic alkalosis suggested primary hyperparathyroidism. However, she was ultimately diagnosed with Bartter syndrome type 2 based on the presence of homozygous pathogenic variation in KCNJ1gene. This is the second reported case of late-onset Bartter syndrome type 2 without hypokalemic alkalosis. Patients with Bartter syndrome may present with high parathyroid hormone levels and hypercalcemia in addition to hypercalciuria. Thus, the present case suggests that the KCNJ1 gene should be included in genetic analysis even in older children with isolated nephrocalcinosis.
Clinical exome sequencing uncovers an unsuspected diagnosis of Bartter syndrome type 2 in a child with incidentally detected nephrocalcinosis.
Nephrocalcinosis is a characteristic feature of both type 1 and type 2 Bartter syndrome. Bartter syndrome type 2 presents antenatally and very early in life. Late-onset presentation with isolated nephrocalcinosis is extremely rare. We describe an 11-year-old girl with incidentally detected medullary nephrocalcinosis on renal ultrasonography. She was clinically suspected to have primary hyperoxaluria based on high urine oxalate. However, clinical exome sequencing revealed a pathogenic missense variant in the KCNJ1 gene leading to the molecular diagnosis of Bartter syndrome type 2. Both parents were heterozygous carriers of the same variant. Subsequent investigations did reveal a mild Bartter syndrome phenotype with mild metabolic alkalosis, high urine chloride and high renin and aldosterone. Our case illustrates phenotypic heterogeneity of Bartter syndrome type 2 and the usefulness of genetic testing in establishing the correct diagnosis and guiding further management in such cases.
Antenatal Bartter syndrome: a new compound heterozygous mutation in exon 2 of KCNJ1 gene.
A 30+6/7-week infant was born by vaginal delivery to a 21-year-old primigravida with pregnancy complicated by polyhydramnios. The infant developed polyuria and significant weight loss in the first 2 weeks of life despite appropriate fluid management. He developed hyponatraemia, hypochloraemia, transient hyperkalaemia and prerenal azotaemia with metabolic acidosis. On further evaluation, he had elevated plasma renin and aldosterone levels. Bartter syndrome was considered in the differential diagnosis. Bartter syndrome gene panel revealed a rare compound heterozygous mutation in exon 2 of the KCNJ1 gene (Lys186Glu/Thr71Met), suggesting antenatal Bartter syndrome (type 2). The infant developed late-onset hypokalaemia and metabolic alkalosis by week 4 of life. He regained birth weight by week 3 of life but failed to thrive (10-20 g/kg/day) despite high caloric intake (140 kcal/kg/day). His electrolyte abnormalities gradually improved, and he was discharged home without the need for electrolyte supplements or medications.
Eight novel KCNJ1 variants and parathyroid hormone overaction or resistance in 5 probands with Bartter syndrome type 2.
Bartter syndrome type 2 (BS2) is an autosomal recessive renal tubular disorder, which is caused by the mutations in KCNJ1. This study was designed to analyze and describe the genotype and clinical features of five Chinese probands with BS2. Identify KCNJ1 gene variants by the next generation sequencing and evaluate their mutation effects according to 2015 American College of Medical Genetics and Genomics (ACMG) standards and guidelines. Ten variants including eight novel ones of KCNJ1 gene were found, the most common type was missense variant. The common symptoms and signs from high to low incidence were: polydipsia and polyuria (5/5), one of them (1/5) presented with diabetes insipidus; maternal polyhydramnios and premature delivery (4/5); growth retardation (3/5). Two patients presented with hypochloremic metabolic alkalosis and hypokalemia; whereas the acid-base disturbance was absent in the others. One patient had evident parathyroid hormone (PTH) resistance (hypocalcemia, hyperphosphatemia and markedly elevated PTH levels), three presented with PTH overacting (hypercalcemia, hypophosphatemia and mild elevated PTH levels), and one showed normal blood calcium and phosphorus concentrations with high-normal PTH levels. All patients had nephrocalcinosis and/or hypercalciuria, and one of them complicated with nephrolithiasis. Indomethacin has significant therapeutic effect on the growth retardation, polydipsia and polyuria and treatment was associated with a decrease in urine calcium excretion, normalization of electrolyte disturbance and PTH parameters. Ten variants of KCNJ1 gene were identified in five Chinese probands. These patients had atypical BS phenotype lacking evident metabolic alkalosis and/or manifesting with PTH overaction/resistance, which reminds clinicians to carefully differentiate BS2 with other parathyroid disorders. This is the first report of BS2 from Chinese populations.
Publicações recentes
Phenotypes and the Importance of Genetic Analysis in Adult Patients with Nephrolithiasis and/or Nephrocalcinosis: A Single-Center Experience.
Late onset Bartter syndrome: Bartter syndrome type 2 presenting with isolated nephrocalcinosis and high parathyroid hormone levels mimicking primary hyperparathyroidism.
Clinical exome sequencing uncovers an unsuspected diagnosis of Bartter syndrome type 2 in a child with incidentally detected nephrocalcinosis.
Antenatal Bartter syndrome: a new compound heterozygous mutation in exon 2 of KCNJ1 gene.
Eight novel KCNJ1 variants and parathyroid hormone overaction or resistance in 5 probands with Bartter syndrome type 2.
📚 EuropePMC566 artigos no totalmostrando 7
Phenotypes and the Importance of Genetic Analysis in Adult Patients with Nephrolithiasis and/or Nephrocalcinosis: A Single-Center Experience.
GenesLate onset Bartter syndrome: Bartter syndrome type 2 presenting with isolated nephrocalcinosis and high parathyroid hormone levels mimicking primary hyperparathyroidism.
Journal of pediatric endocrinology & metabolism : JPEMClinical exome sequencing uncovers an unsuspected diagnosis of Bartter syndrome type 2 in a child with incidentally detected nephrocalcinosis.
CEN case reportsAntenatal Bartter syndrome: a new compound heterozygous mutation in exon 2 of KCNJ1 gene.
BMJ case reportsEight novel KCNJ1 variants and parathyroid hormone overaction or resistance in 5 probands with Bartter syndrome type 2.
Clinica chimica acta; international journal of clinical chemistryThe Urinary Excretion of Uromodulin is Regulated by the Potassium Channel ROMK.
Scientific reports[Gene analysis in a family with adult onset Bartter syndrome type 2].
Zhonghua nei ke za zhiAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Bartter tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Bartter tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Phenotypes and the Importance of Genetic Analysis in Adult Patients with Nephrolithiasis and/or Nephrocalcinosis: A Single-Center Experience.
- Late onset Bartter syndrome: Bartter syndrome type 2 presenting with isolated nephrocalcinosis and high parathyroid hormone levels mimicking primary hyperparathyroidism.
- Clinical exome sequencing uncovers an unsuspected diagnosis of Bartter syndrome type 2 in a child with incidentally detected nephrocalcinosis.
- Antenatal Bartter syndrome: a new compound heterozygous mutation in exon 2 of KCNJ1 gene.
- Eight novel KCNJ1 variants and parathyroid hormone overaction or resistance in 5 probands with Bartter syndrome type 2.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:620220(Orphanet)
- OMIM OMIM:241200(OMIM)
- MONDO:0009424(MONDO)
- GARD:22483(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Bartter tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)