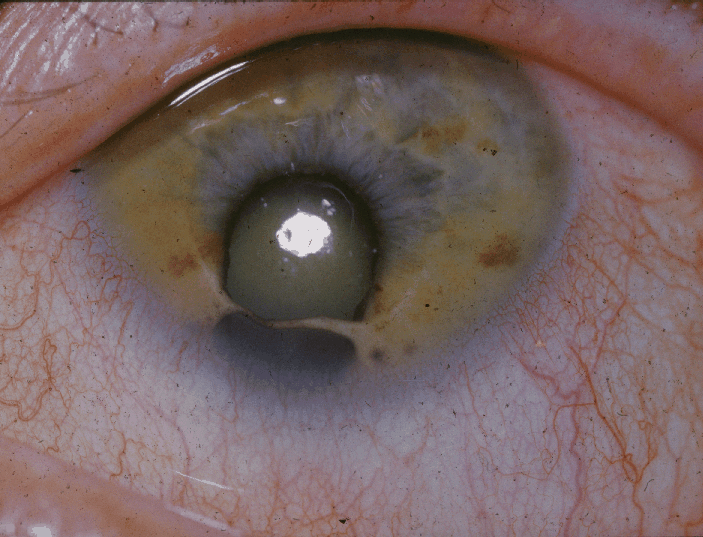
A Síndrome do Olho de Gato (CES) é uma condição cromossômica rara, que se manifesta de formas muito variadas em cada pessoa. A maioria dos pacientes apresenta múltiplas malformações que afetam os olhos (como um defeito na íris, a parte colorida do olho), as orelhas (com pequenos orifícios ou marcas de pele na frente), a região anal (como o ânus fechado ou malformado), o coração e os rins. A deficiência intelectual geralmente é leve ou está no limite do que é considerado normal.
Introdução
O que você precisa saber de cara
A Síndrome do Olho de Gato (CES) é uma condição cromossômica rara, que se manifesta de formas muito variadas em cada pessoa. A maioria dos pacientes apresenta múltiplas malformações que afetam os olhos (como um defeito na íris, a parte colorida do olho), as orelhas (com pequenos orifícios ou marcas de pele na frente), a região anal (como o ânus fechado ou malformado), o coração e os rins. A deficiência intelectual geralmente é leve ou está no limite do que é considerado normal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 14 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome "cat-eye"
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Overexpression of cat eye syndrome chromosome region, candidate 2 in esophageal squamous carcinoma cell promotes tumor aggressiveness by facilitating NF-κB signaling and inhibition of p53-associated apoptosis.
A chromatin remodeling-related gene, cat eye syndrome chromosome region, candidate 2 (CECR2) was identified as candidate gene associated with aggressive phenotypes of esophageal squamous cell carcinoma (ESCC) in a transcriptome analysis. Here, we aimed to elucidate its role and potential for clinical application. We evaluated ESCC cell lines modulating CECR2 expression in vitro. Signaling analysis and inhibitor experiments were conducted to reveal its potential mechanism. Mouse subcutaneous models were established to confirm the effect of knockdown and inhibitor. Expressions of CECR2 on mRNA level were analyzed by qPCR and protein level by TMA, respectively, in two cohorts. CECR2 was significantly upregulated in cancer tissue compared to normal tissue. CECR2 knockdown suppressed metastasis-related biological functions of ESCC cells and increased the sensitivity to principal anticancer reagents for treatment of ESCC, 5-FU and cisplatin. In addition, forced overexpression of CECR2 enhanced proliferation of low-CECR2-expressed cell line. Mechanistically, CECR2 upregulates NF-κB signaling, downregulates acetylated p53 expressions, and activates AKT signaling to enhance NF-κB signaling. Pharmacological inhibition of CECR2 by NVS-CECR2-1 induced cell apoptosis. In mouse subcutaneous models, permanent knockdown mediated by shCECR2 significantly inhibited tumor growth compared to shControl group; tumor growth was inhibited by a continual cycle of intraperitoneal administration of NVS-CECR2-1 compared to PBS only group. Analysis of two cohorts both demonstrated a significant association between high CECR2 mRNA/protein expression levels and poor prognosis. Upregulation of CECR2 in ESCC promotes tumor aggressiveness and may serve as a potential therapeutic target for the treatment of ESCC.
Treatment of bilateral infantile-onset glaucoma associated with Cat Eye Syndrome.
Cat Eye Syndrome Chromosome Region Candidate 2 (CECR2) in chromatin remodeling and cancer: A review.
CECR2 is a bromodomain-containing epigenetic regulator essential for maintaining chromatin architecture and is implicated in diverse biological processes, including cellular development, spermatogenesis, neurulation, and DNA damage response. Acting as a histone acetyl-lysine reader, CECR2 participates in chromatin remodeling through the formation of tissue-specific complexes with ISWI proteins. With its well-established roles in somatic cell reprogramming, CECR2 has recently emerged as a promising therapeutic target in breast cancer metastasis. In this review, we discuss its origin, structural features, regulatory mechanisms, and assembly into multiple protein complexes that mediate its tissue-specific functions in coordination with various transcription factors. Furthermore, we emphasize its significance in pathological conditions such as developmental disorders and cancer, aiming to elucidate the molecular connections that underlie its role in tumor progression and potential as a drug target.
Diagnostic contribution of conventional and molecular karyotyping in congenital diaphragmatic hernia related copy number variations.
Congenital Diaphragmatic Hernia (CDH) results from defects in the developing diaphragm and is characterized by herniation of abdominal contents into the thoracic cavity. Notably, CDH is linked to elevated morbidity and mortality rates due to its association with pulmonary hypoplasia. Copy number variations (CNVs) are significant contributors to the etiology of CDH. We aimed to investigate the involvement of new candidate CNVs in CDH etiology and the effectiveness of karyotyping and array-based Comparative Genomic Hybridization (a-CGH) in CDH diagnosis. Among the 10,536 prenatal cases, 198 cases with CDH were enrolled in this study. Statistical analyses were performed to investigate the possible correlation between CDH type, maternal age, and chromosomal anomaly ratio. Chromosomal analysis was conducted on 188 cases with appropriate material. Consequently, an a-CGH study was executed on 90 cases with normal karyotype results and high-quality DNA material. Chromosomal anomaly frequency was significantly higher (p = 0.0001) in complex than in isolated CDH. In 13.3 % of the cases, various chromosomal anomalies including triploidy, aneuploidy, and those indicating certain syndromes such as Pallister-Killian, Cat-Eye, Turner, and Klinefelter were detected with karyotyping. In nine cases, three pathogenic CNVs including 17q12 microdeletion, 15q11.2 microdeletion, Xq27.1 microduplication, and seven additional CNVs with unknown significance, were identified with the a-CGH study. Our study results significantly support the involvement of chromosomal anomalies and CNVs in CDH etiology. Moreover, our findings revealed new candidate regions for CDH and strengthened the CDH correlation of known CNVs, which may provide a resource for future studies.
Syndromic variants of biliary atresia.
Biliary atresia (BA) may be characterized as an obliterative cholangiopathy presenting in the newborn period with conjugated jaundice, pale stools, and dark urine. It is usually thought of as an isolated anomaly in otherwise normal infants. However, in a minority, other anomalies may be present, some as defined syndromes, others as a non-random association. The most fully characterized is that of the biliary atresia splenic malformation syndrome seen in about 10% of European and North American series with a typical array of unusual extrahepatic anomalies (e.g., situs inversus, polysplenia, absence of the inferior vena cava, and a preduodenal portal vein). Its underlying genetic background is obscure in most cases. There are other syndromes with a definite link to BA, such as Cat-Eye syndrome and Kabuki syndrome, and still others that may have a link, such as Zimmerman-Laband syndrome. Cat eye syndrome (CES), also known as Schmid-Fraccaro syndrome, is a rare genetic disorder named for the vertical iris coloboma observed in some affected individuals. The condition is classically characterized by a triad of features—iris coloboma, anal atresia, and preauricular pits or tags. However, CES can also involve a range of abnormalities affecting the neurodevelopmental, ocular, auricular, nasal, cardiovascular, gastrointestinal, and urogenital systems (see Image. Schematic Diagram Showing Iris Coloboma in Cat Eye Syndrome). The clinical presentation of CES is variable, with differences in affected organ systems, prognosis, genetics, and heritability among individuals. CES is a rare chromosomal disorder first described in the early 1960s by Schmid and Fraccaro. This condition is characterized by a partial tetrasomy or trisomy of chromosome 22q11.1-q11.2. The name "cat eye" originates from ocular colobomas—iris defects—present in about half of affected individuals, which give the pupil a distinctive keyhole or cat-eye appearance. Although ocular coloboma is the eponymous hallmark, CES is fundamentally a multisystem genomic disorder with highly variable expressivity, spanning a spectrum from nearly asymptomatic to severe anomalies across ocular, cardiac, renal, gastrointestinal, skeletal, and neurodevelopmental domains. The association between ocular coloboma and anal atresia was first described by Haab in 1878. The genetic alteration is due to a small supernumerary marker chromosome (sSMC), which was first described in 1965. Schachenmann et al reported 3 pediatric patients and 1 patient’s mother who carried an additional, abnormally small chromosome featuring a submedian centromere, while the rest of the karyotype appeared normal. This sSMC contains the CES critical region (CESCR), located within the proximal portion of chromosome 22q11.2, between the centromere and the LCR22-A region. Additional genetic conditions related to chromosome 22 include the oculo-auriculo-vertebral spectrum (OAVS), DiGeorge syndrome, and mosaic trisomy 22. At the genetic level, CES arises from a supernumerary marker chromosome, often dicentric, composed of material from chromosome 22. In approximately 90% of cases, this marker contains 2 extra copies of the proximal 22q11 region (tetrasomy), while a smaller proportion exhibits an additional copy (trisomy). The critical region encompasses approximately 1.5 to 2 Mb and includes multiple dosage-sensitive genes whose overexpression is believed to contribute to the diverse phenotypic features of CES. Molecular cytogenetic techniques, such as fluorescence in situ hybridization (FISH), array comparative genomic hybridization (aCGH), and, more recently, genome-wide single-nucleotide polymorphism (SNP) microarrays, have replaced traditional karyotyping for precise delineation of the supernumerary chromosome and identification of the breakpoints. This high-resolution genomic mapping is crucial for definitive diagnosis, genotype-phenotype correlations, and recurrence-risk counseling. Clinically, CES is remarkably heterogeneous. The classic triad comprises iris coloboma, preauricular skin tags or pits, and anal atresia or other anorectal malformations. However, no single feature is universally present. Iris coloboma appears in 40% to 60% of cases, preauricular anomalies in up to 70%, and anorectal malformations in about 30% to 50%. Cardiac defects, most commonly total or partial atrioventricular septal defects and tetralogy of Fallot, occur in approximately half of patients and are a major contributor to early morbidity and mortality. Renal anomalies, reported in 20% to 40% of cases, may include unilateral renal agenesis, duplex collecting systems, hydronephrosis, and vesicoureteral reflux. Skeletal abnormalities range from vertebral segmentation defects to limb anomalies. Otolaryngological manifestations may include hearing loss from middle-ear dysplasia. Less commonly, gastrointestinal anomalies beyond anorectal malformations—such as duodenal atresia or Hirschsprung disease—have also been documented. Neurodevelopmental outcomes in CES vary widely. Although some children achieve developmental milestones within normal limits, others present with global developmental delay, intellectual disability, or features consistent with autism spectrum disorder. Hypotonia during infancy and feeding difficulties—often related to underlying gastrointestinal anomalies—may further compromise early growth. Growth parameters can be affected, with some patients exhibiting short stature or failure to thrive; however, many ultimately achieve normal height and weight. Behavioral phenotypes—such as attention-deficit/hyperactivity disorder (ADHD) and anxiety disorders—have also been reported, emphasizing the importance of comprehensive developmental and psychological assessment. Ophthalmic manifestations in CES extend beyond the iris coloboma. Additional anomalies such as chorioretinal colobomas, microphthalmia, cataracts, microcornea, and strabismus can contribute to visual impairment. A comprehensive ophthalmologic evaluation includes slit-lamp biomicroscopy to assess anterior segment abnormalities, indirect ophthalmoscopy for posterior segment examination, and optical coherence tomography (OCT), when available, to delineate the extent of colobomatous defects. Early detection and management of refractive errors, amblyopia, and strabismus are essential to support optimal visual development. Surgical intervention for coloboma is rarely indicated and is typically reserved for cases involving severe aniridia-like photophobia or significant cosmetic concerns (see Image. Schematic Diagram Showing Chorioretinal Coloboma in Cat Eye Syndrome). The management of CES depends on the organ systems involved and the severity of associated malformations. Given the significant clinical heterogeneity, an individualized, interprofessional approach is essential. This activity outlines the genetic and phenotypic spectrum of CES and outlines strategies for tailoring medical care to each patient’s needs. Cardiac evaluation at diagnosis is mandatory. Echocardiography within the first weeks of life is essential for detecting structural heart disease; in moderate-to-severe cases, surgical repair during infancy may be lifesaving. Long-term cardiology follow-up is critical to monitor for residual defects, arrhythmias, and pulmonary hypertension. Similarly, early renal ultrasonography is recommended to identify anatomical anomalies, guide urologic management, and prevent complications such as hypertension or renal insufficiency. Gastroenterological and colorectal management primarily focuses on anorectal malformations. Posterior sagittal anorectoplasty (PSARP) is the standard repair for imperforate anus, with timing and technical details tailored to the patient’s specific anatomy and overall health. Nutritional support—ranging from gavage or gastrostomy feeding in neonates to dietary modifications in older children—is essential, especially when gastrointestinal motility disorders or malabsorption are present. Audiologic and otologic care begins with newborn hearing screening. Conductive hearing loss due to middle ear anomalies may require interventions such as tympanostomy tubes or myringotomy. Speech therapy and educational support, tailored to the child’s developmental needs, are essential for optimizing communication outcomes. Genetic counseling for families includes discussion of recurrence risk, which is generally low (<1%) in de novo cases but higher in familial instances when a parent carries the small supernumerary marker chromosome in a balanced form. Secondary complications may include endocrine disorders, particularly growth hormone deficiency and thyroid dysfunction, necessitating regular endocrinologic screening. Orthopedic evaluations focus on detecting scoliosis and limb-length discrepancies. Dental and orthodontic assessments help identify malocclusion and enamel hypoplasia. Psychosocial support for families—including referrals to patient advocacy groups and peer support networks—promotes coping strategies and shared experiences. From a research perspective, CES provides valuable insights into gene dosage effects in contiguous-gene syndromes. The 22q11 region implicated in CES overlaps with that of DiGeorge syndrome (22q11.2 deletion), yet their phenotypes differ, reflecting divergent consequences of haploinsufficiency versus gene overexpression. Current studies focus on elucidating the roles of candidate genes such as CECR1 (which encodes adenosine deaminase 2) and CECR2 (involved in chromatin remodeling) in contributing to CES manifestations. Animal models with targeted duplications of the 22q11 region are under development to investigate relevant developmental pathways. Additionally, next-generation sequencing techniques show promise in detecting cryptic rearrangements and refining genotype–phenotype correlations, ultimately improving prognostic accuracy and identifying potential therapeutic targets. In summary, CES is a complex, multisystem chromosomal disorder. While its hallmark features include ocular coloboma, ear anomalies, and anorectal malformations, the condition also encompasses a wider phenotypic spectrum affecting the cardiac, renal, skeletal, neurodevelopmental, and endocrine systems. Accurate diagnosis relies on high-resolution cytogenetic and molecular techniques. Effective management requires coordinated multidisciplinary care, involving specialties from neonatology and cardiology to ophthalmology, urology, and developmental pediatrics. As advances in molecular genetics continue, they will enhance personalized prognostic counseling and enable the development of targeted therapies, ultimately improving outcomes for individuals and families affected by this rare but informative genomic syndrome.
Publicações recentes
Heterozygous CECR2 Variants Support a Distinct Neurodevelopmental Syndrome with Features Overlapping Cat Eye Syndrome.
Treatment of bilateral infantile-onset glaucoma associated with Cat Eye Syndrome.
Cat Eye Syndrome Chromosome Region Candidate 2 (CECR2) in chromatin remodeling and cancer: A review.
Overexpression of cat eye syndrome chromosome region, candidate 2 in esophageal squamous carcinoma cell promotes tumor aggressiveness by facilitating NF-κB signaling and inhibition of p53-associated apoptosis.
🥈 ObservacionalDiagnostic contribution of conventional and molecular karyotyping in congenital diaphragmatic hernia related copy number variations.
📚 EuropePMC136 artigos no totalmostrando 84
Treatment of bilateral infantile-onset glaucoma associated with Cat Eye Syndrome.
American journal of ophthalmology case reportsCat Eye Syndrome Chromosome Region Candidate 2 (CECR2) in chromatin remodeling and cancer: A review.
International journal of biological macromoleculesOverexpression of cat eye syndrome chromosome region, candidate 2 in esophageal squamous carcinoma cell promotes tumor aggressiveness by facilitating NF-κB signaling and inhibition of p53-associated apoptosis.
Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of MexicoDiagnostic contribution of conventional and molecular karyotyping in congenital diaphragmatic hernia related copy number variations.
Taiwanese journal of obstetrics & gynecologySyndromic variants of biliary atresia.
World journal of pediatric surgeryUpdates in Biliary Atresia: Aetiology, Diagnosis and Surgery.
Children (Basel, Switzerland)The CECR2 bromodomain displays distinct binding modes to select for acetylated histone proteins versus non-histone ligands.
bioRxiv : the preprint server for biologyClinical and molecular cytogenetic findings of cat eye syndrome and a 2-year-old patient with congenital aural atresia and hearing loss.
BMC pediatricsMortality in Patients with 22q11.2 Rearrangements.
GenesPrenatal diagnosis and genetic analysis of small supernumerary marker chromosomes in the eastern chinese han population: A retrospective study of 36 cases.
Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biologyHuman Genetics of Tetralogy of Fallot and Double-Outlet Right Ventricle.
Advances in experimental medicine and biologyCD138 as a Specific CSF Biomarker of Multiple Sclerosis.
Neurology(R) neuroimmunology & neuroinflammationOptical controlled and nuclear targeted CECR2 competitor to downregulate CSF-1 for metastatic breast cancer immunotherapy.
BiomaterialsCat eye syndrome: Clinical, cytogenetics and familial findings in a large cohort of 43 patients highlighting the importance of congenital heart disease and inherited cases.
American journal of medical genetics. Part ACat eye syndrome caused by 22q11.1q11.21 duplication: case report in a Chinese family.
Molecular cytogeneticsLncRNA CECR7 boosts hepatocellular carcinoma progression by recruiting RNA binding protein U2AF2 to enhance the stability of EXO1 mRNA.
HeliyonVasculitis associated with adenosine deaminase 2 deficiency: at the crossroads between Behçet's disease and autoinflammation. A viewpoint.
ReumatismoA new CECR1 mutation associated with severe hematological involvement in ADA2 deficiency.
Immunity, inflammation and diseaseOverlapping Spectrum of Craniofacial Microsomia Phenotype in Cat-Eye Syndrome.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationCat Eye Syndrome with a Unique Liver and Dermatological Presentation.
CureusA Chinese family with cat eye syndrome and abnormality of eye movement: First case report.
Frontiers in pediatricsDifferent Types of Deletions Created by Low-Copy Repeats Sequences Location in 22q11.2 Deletion Syndrome: Genotype-Phenotype Correlation.
GenesCongenital hypopituitarism and multiple midline defects in a newborn with non-familial Cat Eye syndrome.
Italian journal of pediatricsCat-Eye Syndrome: A Report of Two Cases and Literature Review.
CureusChromosomal Abnormalities and Pregnancy Outcomes for Fetuses With Gastrointestinal Tract Obstructions.
Frontiers in pediatricsThe Role of Repeat DNA Sequences in Human Evolution and Disease.
Journal of the Association of Genetic TechnologistsDiscovery of a highly potent CECR2 bromodomain inhibitor with 7H-pyrrolo[2,3-d] pyrimidine scaffold.
Bioorganic chemistryCECR2 drives breast cancer metastasis by promoting NF-κB signaling and macrophage-mediated immune suppression.
Science translational medicineBiliary Atresia: Clinical Phenotypes and Aetiological Heterogeneity.
Journal of clinical medicineAdenosine deaminase 2 produced by infiltrative monocytes promotes liver fibrosis in nonalcoholic fatty liver disease.
Cell reportsCongenital Eyelid Imbrication and Floppy Eyelid Syndrome in a Patient With Cat Eye Syndrome.
Ophthalmic plastic and reconstructive surgeryA Novel LC-MS/MS-Based Method for the Diagnosis of ADA2 Deficiency from Dried Plasma Spot.
Molecules (Basel, Switzerland)A child with cat-eye syndrome and oculo-auriculo-vertebral spectrum phenotype: A discussion around molecular cytogenetic findings.
European journal of medical geneticsNext-generation phenotyping in cat-eye syndrome based on computer-aided facial dysmorphology analysis of normal photographs.
Molecular genetics & genomic medicineA Novel Variant of Adenosine Deaminase 2 Deficiency Presented With Chronic Thrombocytopenia, Anemia, and Early-Onset Stroke.
CureusThe role of CECR1 in the immune-modulatory effects of butyrate and correlation between ADA2 and M1/M2 chemokines in tuberculous pleural effusion.
International immunopharmacologyA De Novo sSMC (22) Characterized by High-Resolution Chromosome Microarray Analysis in a Chinese Boy with Cat-Eye Syndrome.
Case reports in geneticsAn Unusual Association: Total Anomalous Pulmonary Venous Return and Aortic Arch Obstruction in Patients with Cat Eye Syndrome.
Journal of pediatric geneticsCecr2 mutant mice as a model for human cat eye syndrome.
Scientific reportsDetailed analysis of Japanese patients with adenosine deaminase 2 deficiency reveals characteristic elevation of type II interferon signature and STAT1 hyperactivation.
The Journal of allergy and clinical immunologyMosaic cat eye syndrome in a child with unilateral iris coloboma.
Ophthalmic geneticsMultimodal Imaging of Large Optic Disc Coloboma: A Report of Three Cases.
Case reports in ophthalmologyPrenatal diagnosis and molecular cytogenetic identification of small supernumerary marker chromosomes: analysis of three prenatal cases using chromosome microarray analysis.
AgingCytotoxic activity of bromodomain inhibitor NVS-CECR2-1 on human cancer cells.
Scientific reportsAtypical presentation of Cat Eye Syndrome in an infant with Peters anomaly and microphthalmia with cyst.
Ophthalmic geneticsCardiac-associated biliary atresia (CABA): a prognostic subgroup.
Archives of disease in childhoodOptimization of Potent ATAD2 and CECR2 Bromodomain Inhibitors with an Atypical Binding Mode.
Journal of medicinal chemistryAtypical phenotype of an old disease or typical phenotype of a new disease: deficiency of adenosine deaminase 2.
The Turkish journal of pediatrics[Cat-eye syndrome (a psychiatric aspect)].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaRare otologic presentation of cat eye syndrome.
Annals of Saudi medicineMosaicism: Reason for Normal Phenotypes in Carriers of Small Supernumerary Marker Chromosomes With Known Adverse Outcome. A Systematic Review.
Frontiers in geneticsA Child Diagnosed With Treatment-Resistant Polyarteritis Nodosa: Can the Clinical Diagnosis Be Different?
Archives of rheumatologyConcurrent Hirschsprung's disease and anorectal malformation: a systematic review.
Pediatric surgery internationalThe same mutation in a family with adenosine deaminase 2 deficiency.
Rheumatology internationalFree episomal and integrated HBV DNA in HBsAg-negative patients with intrahepatic cholangiocarcinoma.
Oncotarget[Prenatal diagnosis and clinical analysis of two fetuses with Cat-eye syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsDe novo Unbalanced 1;22 Translocation with 22q11 Deletion Syndrome.
Cytogenetic and genome researchALPS-Like Phenotype Caused by ADA2 Deficiency Rescued by Allogeneic Hematopoietic Stem Cell Transplantation.
Frontiers in immunologyAdenosine Deaminase Two and Immunoglobulin M Accurately Differentiate Adult Sneddon's Syndrome of Unknown Cause.
Cerebrovascular diseases (Basel, Switzerland)A 9.5-year-old boy with recurrent neurological manifestations and severe hypertension, treated initially for polyarteritis nodosa, was subsequently diagnosed with adenosine deaminase type 2 deficiency (DADA2) which responded to anti-TNF-α.
Paediatrics and international child healthRetrospectively investigating the 12-year experience of prenatal diagnosis of small supernumerary marker chromosomes through array comparative genomic hybridization.
Taiwanese journal of obstetrics & gynecologyA Chinese DADA2 patient: report of two novel mutations and successful HSCT.
ImmunogeneticsComparative proteomic analysis of cat eye syndrome critical region protein 1- function in tumor-associated macrophages and immune response regulation of glial tumors.
OncotargetCongenital diaphragmatic hernia in a case of Cat eye syndrome.
Clinical case reportsDeficiency of Adenosine Deaminase 2 in Adult Siblings: Many Years of a Misdiagnosed Disease With Severe Consequences.
Frontiers in immunology[Cytogenetic and molecular genetic analysis of the amniotic fluid cells of a fetus with pseudodicentric isochromosome 22 resulting in partial tetraploidy of 22q].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsIncomplete penetrance, variable expressivity, or dosage insensitivity in four families with directly transmitted unbalanced chromosome abnormalities.
American journal of medical genetics. Part AMüllerian Agenesis in Cat Eye Syndrome and 22q11 Chromosome Abnormalities: A Case Report and Literature Review.
Journal of pediatric and adolescent gynecologyGNE-886: A Potent and Selective Inhibitor of the Cat Eye Syndrome Chromosome Region Candidate 2 Bromodomain (CECR2).
ACS medicinal chemistry lettersNext generation phenotyping in Emanuel and Pallister-Killian syndrome using computer-aided facial dysmorphology analysis of 2D photos.
Clinical geneticsCECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma.
OncogeneActivation of CECR1 in M2-like TAMs promotes paracrine stimulation-mediated glial tumor progression.
Neuro-oncologyRefractory Pure Red Cell Aplasia Manifesting as Deficiency of Adenosine Deaminase 2.
Journal of pediatric hematology/oncologyMycobacterium tuberculosis ESAT6 and CPF10 Induce Adenosine Deaminase 2 mRNA Expression in Monocyte-Derived Macrophages.
Tuberculosis and respiratory diseasesStroke as Initial Manifestation of Adenosine Deaminase 2 Deficiency.
NeuropediatricsMonogenic polyarteritis: the lesson of ADA2 deficiency.
Pediatric rheumatology online journalCNV analysis in 169 patients with bladder exstrophy-epispadias complex.
BMC medical geneticsDeficiency of Adenosine Deaminase Type 2: A Description of Phenotype and Genotype in Fifteen Cases.
Arthritis & rheumatology (Hoboken, N.J.)Proptosis, Micrognathia, Low Set Ear and Chest Deformity in a Patient with Extra Marker Chromosome 22.
Acta medica IranicaAnalysis of chromosome 22q11 copy number variations by multiplex ligation-dependent probe amplification for prenatal diagnosis of congenital heart defect.
Molecular cytogeneticsGrowth hormone deficiency and pituitary malformation in a recurrent Cat-Eye syndrome: a family report.
Annales d'endocrinologieQuadricuspid aortic valve and anomalous systemic venous connection in a patient with cat-eye syndrome.
CirculationType B Interrupted Aortic Arch and Hydrocephalus Associated with Mosaicism of a 1.37 Mb Amplified Cat Eye Syndrome Critical Region.
Pediatrics and neonatologySevere psychomotor delay in a severe presentation of cat-eye syndrome.
Case reports in geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome "cat-eye".
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome "cat-eye"
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Overexpression of cat eye syndrome chromosome region, candidate 2 in esophageal squamous carcinoma cell promotes tumor aggressiveness by facilitating NF-κB signaling and inhibition of p53-associated apoptosis.Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico· 2026· PMID 40760241mais citado
- Treatment of bilateral infantile-onset glaucoma associated with Cat Eye Syndrome.
- Cat Eye Syndrome Chromosome Region Candidate 2 (CECR2) in chromatin remodeling and cancer: A review.
- Diagnostic contribution of conventional and molecular karyotyping in congenital diaphragmatic hernia related copy number variations.
- Syndromic variants of biliary atresia.
- Heterozygous CECR2 Variants Support a Distinct Neurodevelopmental Syndrome with Features Overlapping Cat Eye Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:195(Orphanet)
- OMIM OMIM:115470(OMIM)
- MONDO:0007276(MONDO)
- GARD:26(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1199621(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome "cat-eye"
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata