A síndrome coração-mão tipo 2 é uma síndrome coração-mão extremamente rara descrita em duas famílias até o momento, que se caracteriza por malformações dos membros superiores (braquitelefalangia tipo D, deltóides hipoplásicos, leve encurtamento do quarto e quinto metacarpos em alguns indivíduos, anomalias esqueléticas no úmero, rádio, ulna e ossos tenares) e arritmias cardíacas (ritmos juncionais e fibrilação atrial).
Introdução
O que você precisa saber de cara
A síndrome coração-mão tipo 2 é uma síndrome coração-mão extremamente rara descrita em duas famílias até o momento, que se caracteriza por malformações dos membros superiores (braquitelefalangia tipo D, deltóides hipoplásicos, leve encurtamento do quarto e quinto metacarpos em alguns indivíduos, anomalias esqueléticas no úmero, rádio, ulna e ossos tenares) e arritmias cardíacas (ritmos juncionais e fibrilação atrial).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 15 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 25 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome coração-mão tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Elucidating the roles of SOD3 correlated genes and reactive oxygen species in rare human diseases using a bioinformatic-ontology approach.
Superoxide Dismutase 3 (SOD3) scavenges extracellular superoxide giving a hydrogen peroxide metabolite. Both Reactive Oxygen Species diffuse through aquaporins causing oxidative stress and biomolecular damage. SOD3 is differentially expressed in cancer and this research utilises Gene Expression Omnibus data series GSE2109 with 2,158 cancer samples. Genome-wide expression correlation analysis was conducted with SOD3 as the seed gene. Categorical SOD3 Pearson Correlation gene lists incrementing in correlation strength by 0.01 from ρ≥|0.34| to ρ≥|0.41| were extracted from the data. Positively and negatively SOD3 correlated genes were separated for each list and checked for significance against disease overlapping genes in the ClinVar and Orphanet databases via Enrichr. Disease causal genes were added to the relevant gene list and checked against Gene Ontology, Phenotype Ontology, and Elsevier Pathways via Enrichr before the significant ontologies containing causal and non-overlapping genes were reviewed with a literature search for possible disease and oxidative stress associations. 12 significant individually discriminated disorders were identified: Autosomal Dominant Cutis Laxa (p = 6.05x10-7), Renal Tubular Dysgenesis of Genetic Origin (p = 6.05x10-7), Lethal Arteriopathy Syndrome due to Fibulin-4 Deficiency (p = 6.54x10-9), EMILIN-1-related Connective Tissue Disease (p = 6.54x10-9), Holt-Oram Syndrome (p = 7.72x10-10), Multisystemic Smooth Muscle Dysfunction Syndrome (p = 9.95x10-15), Distal Hereditary Motor Neuropathy type 2 (p = 4.48x10-7), Congenital Glaucoma (p = 5.24x210-9), Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome (p = 3.77x10-16), Classical-like Ehlers-Danlos Syndrome type 1 (p = 3.77x10-16), Retinoblastoma (p = 1.9x10-8), and Lynch Syndrome (p = 5.04x10-9). 35 novel (21 unique) genes across 12 disorders were identified: ADNP, AOC3, CDC42EP2, CHTOP, CNN1, DES, FOXF1, FXR1, HLTF, KCNMB1, MTF2, MYH11, PLN, PNPLA2, REST, SGCA, SORBS1, SYNPO2, TAGLN, WAPL, and ZMYM4. These genes are proffered as potential biomarkers or therapeutic targets for the corresponding rare diseases discussed.
Functional characterization vs in silico prediction for TBX5 missense and splice variants in Holt-Oram syndrome.
Predicting effects of genomic variants has become a real challenge in the diagnosis of rare human diseases. Holt-Oram syndrome is an autosomal condition characterized by the association of radial and heart defects, due to variants in TBX5. Most variants are predicted to be truncating and result in haploinsufficiency. The pathogenicity of missense or splice variants is harder to demonstrate. Fourteen TBX5 variants of uncertain significance (5 missense, 9 splice) and 6 likely pathogenic missense variants were selected for functional testing, depending on the variant-type (immunolocalization, western blot, reporter assays, minigene splice assays, and reverse transcription-polymerase chain reaction). Results were compared with in silico predictions. Functional tests allowed to reclassify 9/14 variants of uncertain significance in TBX5 as likely pathogenic, confirming their role in Holt-Oram syndrome. We demonstrated loss of function (n = 8) or gain of function (n = 1) for 9 of the 11 missense variants, whereas no functional impact was shown for the 2 variants: p.(Gly195Ala) and p.(Ser261Cys), as suggested by contradictory predictions of in silico approaches. Of 9 splice variants predicted to affect splicing by SpliceAI, we observed partial or complete exon skipping (n = 6), intron retention (n = 2) or exon shortening (n = 1), inducing frame shifting with premature stop codons. Bioinformatic and biological approaches are complementary, together with a good knowledge of clinical conditions, for accurate American College of Medical Genetics and Genomics classification in human rare diseases.
Transcriptional regulation of human T-box 5 gene (TBX5) by bone- and cardiac-related transcription factors.
T-box 5 (TBX5) protein belongs to the T-box family whose members play a crucial role in cell-type specification, morphogenesis and organogenesis. TBX5 is a transcription factor important for cardiac development and upper limbs formation and its haploinsufficiency causes Holt-Oram syndrome (HOS). An increase in TBX5 dosage also leads to HOS, suggesting that TBX5 is a dose-sensitive transcription factor that needs to be tightly regulated but the molecular mechanisms involved remain unclear. In this work we report the cloning and functional analysis of human TBX5 promoter region 1 (upstream of exon 1) and promoter region 2 (upstream of exon 2), that probably regulate the transcription of the different transcript variants. In silico analysis showed several binding sites for cardiac and skeletal related transcription factors (TFs) and their functionality was assessed using promoter-luciferase constructions and TF-expressing vectors. MEF2A (Myocyte enhancer factor 2 A) was shown to positively regulate both TBX5 promoters, while EGR1 (early growth response 1) repressed both promoters. SOX9 (SRY (sex determining region Y)-box 9) repressed only the activity of promoter region 2. Interestingly, YY1 (Yin and yang 1) repressed promoter region 1 (that regulates the expression of variant 1 and 3), but activated promoter region 2 (that regulates the expression of variant 4). In conclusion, this work provides novel insights toward the better understanding of TBX5 transcriptional regulation by cardiac- and skeletal-related TFs.
Prevalence and Spectrum of TBX5 Mutation in Patients with Lone Atrial Fibrillation.
Atrial fibrillation (AF), the most common type of cardiac rhythm disturbance encountered in clinical practice, is associated with substantially increased morbidity and mortality. Aggregating evidence demonstrates that abnormal cardiovascular development is involved in the pathogenesis of AF. A recent study has revealed that the TBX5 gene, which encodes a T-box transcription factor key to cardiovascular development, was associated with AF and atypical Holt-Oram syndrome. However, the prevalence and spectrum of TBX5 mutation in patients with lone AF remain unclear. In this study, the coding regions and splicing junction sites of TBX5 were sequenced in 192 unrelated patients with lone AF and 300 unrelated ethnically-matched healthy individuals used as controls. The causative potential of the identified TBX5 variation was evaluated by MutationTaster and PolyPhen-2. The functional effect of the mutant TBX5 was assayed by using a dual-luciferase reporter assay system. As a result, a novel heterozygous TBX5 mutation, p.H170D, was identified in a patient, with a mutational prevalence of approximately 0.52%. This mutation, which was absent in the 300 control individuals, altered the amino acid completely conserved evolutionarily across species, and was predicted to be disease-causing. Functional deciphers showed that the mutant TBX5 was associated with significantly reduced transcriptional activity when compared with its wild-type counterpart. Furthermore, the mutation significantly decreased the synergistic activation between TBX5 and NKX2-5 or GATA4. The findings expand the mutational spectrum of TBX5 linked to AF and provide new evidence that dysfunctional TBX5 may contribute to lone AF.
TBX5 loss-of-function mutation contributes to atrial fibrillation and atypical Holt-Oram syndrome.
Previous genome-wide association studies have demonstrated that single nucleotide polymorphisms in T‑box (TBX)5 are associated with increased susceptibility to atrial fibrillation (AF), and a recent study has causally linked a TBX5 mutation to atypical Holt-Oram syndrome and paroxysmal AF. However, the prevalence and spectrum of TBX5 mutations in patients with AF remain to be elucidated. In the present study, a cohort of 190 unrelated patients with idiopathic AF were prospectively recruited, with 400 unrelated healthy individuals recruited as controls. The coding exons and flanking introns of the TBX5 gene were sequenced in the participants. The functional characteristics of the mutant TBX5 were delineated in contrast with its wild‑type counterpart using a dual‑luciferase reporter assay system. As a result, a novel heterozygous TBX5 mutation, p.P132S, was identified in an index patient with AF, with a mutational prevalence of ~0.53%. Genetic analysis of the proband's family showed that the mutation co‑segregated with AF, and was transmitted in an autosomal dominant pattern. The missense mutation was absent in the 800 control chromosomes, and the altered amino acid was completely evolutionarily conserved across species. Functional analyses revealed that the mutant TBX5 had significantly reduced transcriptional activity. Furthermore, the mutation markedly decreased the synergistic activation between TBX5 and NK2 homeobox 5, another transcription factor which has been causatively linked to AF. The present study was the first, to the best of our knowledge, to report on the association between a TBX5 loss‑of‑function mutation and increased susceptibility to AF. These results provide novel insight into the molecular mechanism underpinning AF, and have potential implications in the development of novel prophylactic and therapeutic strategies for AF, the most common form of sustained cardiac arrhythmia.
Publicações recentes
Unusual metastatic patterns of urologic malignancies: a case series and literature review.
📖 RevisãoHolt-Oram Syndrome With Atrial Septal Defect.
Lateral thinking in syndromic congenital cardiovascular disease.
📚 EuropePMC29 artigos no totalmostrando 6
Elucidating the roles of SOD3 correlated genes and reactive oxygen species in rare human diseases using a bioinformatic-ontology approach.
PloS oneFunctional characterization vs in silico prediction for TBX5 missense and splice variants in Holt-Oram syndrome.
Genetics in medicine : official journal of the American College of Medical GeneticsTranscriptional regulation of human T-box 5 gene (TBX5) by bone- and cardiac-related transcription factors.
GeneTBX5 loss-of-function mutation contributes to atrial fibrillation and atypical Holt-Oram syndrome.
Molecular medicine reportsPrevalence and Spectrum of TBX5 Mutation in Patients with Lone Atrial Fibrillation.
International journal of medical sciencesDefining Features of the Upper Extremity in Holt-Oram Syndrome.
The Journal of hand surgeryAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome coração-mão tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome coração-mão tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Elucidating the roles of SOD3 correlated genes and reactive oxygen species in rare human diseases using a bioinformatic-ontology approach.
- Functional characterization vs in silico prediction for TBX5 missense and splice variants in Holt-Oram syndrome.Genetics in medicine : official journal of the American College of Medical Genetics· 2024· PMID 39268717mais citado
- Transcriptional regulation of human T-box 5 gene (TBX5) by bone- and cardiac-related transcription factors.
- Prevalence and Spectrum of TBX5 Mutation in Patients with Lone Atrial Fibrillation.
- TBX5 loss-of-function mutation contributes to atrial fibrillation and atypical Holt-Oram syndrome.
- Unusual metastatic patterns of urologic malignancies: a case series and literature review.
- Holt-Oram Syndrome.
- Holt-Oram Syndrome With Atrial Septal Defect.
- Holt-Oram Syndrome.
- Lateral thinking in syndromic congenital cardiovascular disease.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1350(Orphanet)
- MONDO:0015284(MONDO)
- GARD:9847(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q4892313(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
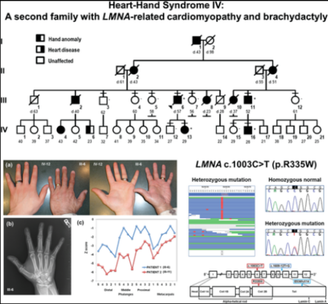
Síndrome coração-mão tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata