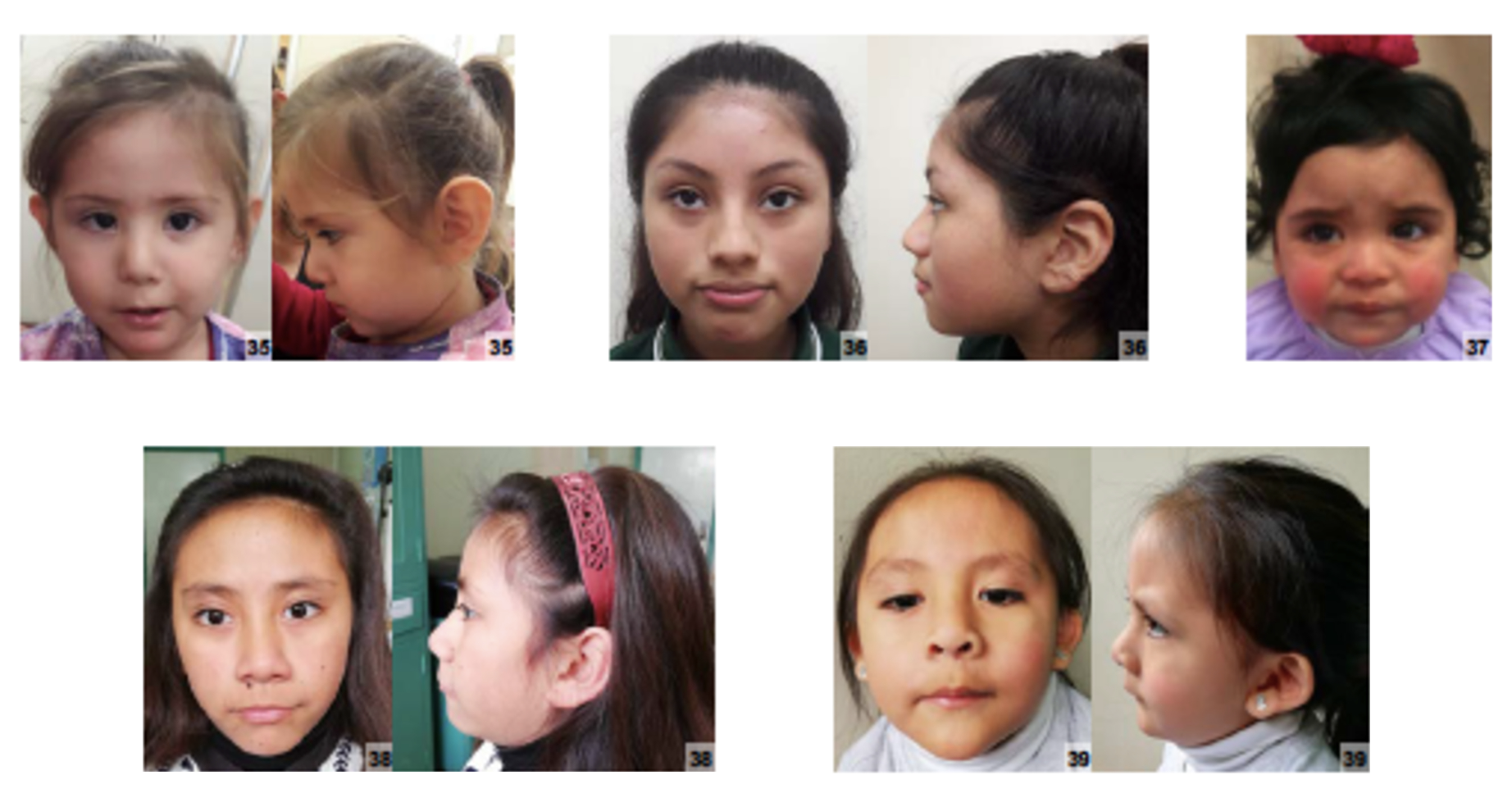
A síndrome do couro cabeludo-orelha-mamilo é caracterizada por três características principais: áreas de pele sensível, avermelhada e sem pelos no couro cabeludo (presentes ao nascer e que cicatrizam durante a infância); orelhas que se destacam, mas são pouco desenvolvidas, com a parte externa (pavilhão auditivo) quase ausente; e a ausência das duas mamas (seios). Até o momento, trinta casos foram descritos. Alterações nos rins e no sistema urinário, assim como catarata, também foram observadas. A transmissão é autossômica dominante.
Introdução
O que você precisa saber de cara
A síndrome do couro cabeludo-orelha-mamilo é caracterizada por três características principais: áreas de pele sensível, avermelhada e sem pelos no couro cabeludo (presentes ao nascer e que cicatrizam durante a infância); orelhas que se destacam, mas são pouco desenvolvidas, com a parte externa (pavilhão auditivo) quase ausente; e a ausência das duas mamas (seios). Até o momento, trinta casos foram descritos. Alterações nos rins e no sistema urinário, assim como catarata, também foram observadas. A transmissão é autossômica dominante.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 26 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 71 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisMay repress the transcriptional activity of AP-2 family members, including TFAP2A, TFAP2B and TFAP2C to various extent
Nucleus
Scalp-ear-nipple syndrome
A disease characterized by aplasia cutis congenita of the scalp, breast anomalies that range from hypothelia or athelia to amastia, and minor anomalies of the external ears. Less frequent clinical characteristics include nail dystrophy, dental anomalies, cutaneous syndactyly of the digits, and renal malformations. Penetrance appears to be high, although there is substantial variable expressivity within families.
Variantes genéticas (ClinVar)
57 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 44 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome couro cabeludo-pavilhão auricular-mamilo
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Scalp-Ear-Nipple Syndrome: A Rare Sporadic Presentation.
A BTB extension and ion-binding domain contribute to the pentameric structure and TFAP2A binding of KCTD1.
KCTD family proteins typically assemble into cullin-RING E3 ligases. KCTD1 is an atypical member that functions instead as a transcriptional repressor. Mutations in KCTD1 cause developmental abnormalities and kidney fibrosis in scalp-ear-nipple syndrome. Here, we present unexpected mechanistic insights from the structure of human KCTD1. Disease-causing mutation P20S maps to an unrecognized extension of the BTB domain that contributes to both its pentameric structure and TFAP2A binding. The C-terminal domain (CTD) shares its fold and pentameric assembly with the GTP cyclohydrolase I feedback regulatory protein (GFRP) despite lacking discernible sequence similarity. Most surprisingly, the KCTD1 CTD establishes a central channel occupied by alternating sodium and iodide ions that restrict TFAP2A dissociation. The elucidation of the structure redefines the KCTD1 BTB domain fold and identifies an unexpected ion-binding site for future study of KCTD1's function in the ectoderm, neural crest, and kidney.
Structural studies of KCTD1 and its disease-causing mutant P20S provide insights into the protein function and misfunction.
Members of the KCTD protein family play key roles in fundamental physio-pathological processes including cancer, neurodevelopmental/neuropsychiatric, and genetic diseases. Here, we report the crystal structure of the KCTD1 P20S mutant, which causes the scalp-ear-nipple syndrome, and molecular dynamics (MD) data on the wild-type protein. Surprisingly, the structure unravels that the N-terminal region, which precedes the BTB domain (preBTB) and bears the disease-associated mutation, adopts a folded polyproline II (PPII) state. The KCTD1 pentamer is characterized by an intricate architecture in which the different subunits mutually exchange domains to generate a closed domain swapping motif. Indeed, the BTB of each chain makes peculiar contacts with the preBTB and the C-terminal domain (CTD) of an adjacent chain. The BTB-preBTB interaction consists of a PPII-PPII recognition motif whereas the BTB-CTD contacts are mediated by an unusual (+/-) helix discontinuous association. The inspection of the protein structure, along with the data emerged from the MD simulations, provides an explanation of the pathogenicity of the P20S mutation and unravels the role of the BTB-preBTB interaction in the insurgence of the disease. Finally, the presence of potassium bound to the central cavity of the CTD pentameric assembly provides insights into the role of KCTD1 in metal homeostasis.
BTB domain mutations perturbing KCTD15 oligomerisation cause a distinctive frontonasal dysplasia syndrome.
KCTD15 encodes an oligomeric BTB domain protein reported to inhibit neural crest formation through repression of Wnt/beta-catenin signalling, as well as transactivation by TFAP2. Heterozygous missense variants in the closely related paralogue KCTD1 cause scalp-ear-nipple syndrome. Exome sequencing was performed on a two-generation family affected by a distinctive phenotype comprising a lipomatous frontonasal malformation, anosmia, cutis aplasia of the scalp and/or sparse hair, and congenital heart disease. Identification of a de novo missense substitution within KCTD15 led to targeted sequencing of DNA from a similarly affected sporadic patient, revealing a different missense mutation. Structural and biophysical analyses were performed to assess the effects of both amino acid substitutions on the KCTD15 protein. A heterozygous c.310G>C variant encoding p.(Asp104His) within the BTB domain of KCTD15 was identified in an affected father and daughter and segregated with the phenotype. In the sporadically affected patient, a de novo heterozygous c.263G>A variant encoding p.(Gly88Asp) was present in KCTD15. Both substitutions were found to perturb the pentameric assembly of the BTB domain. A crystal structure of the BTB domain variant p.(Gly88Asp) revealed a closed hexameric assembly, whereas biophysical analyses showed that the p.(Asp104His) substitution resulted in a monomeric BTB domain likely to be partially unfolded at physiological temperatures. BTB domain substitutions in KCTD1 and KCTD15 cause clinically overlapping phenotypes involving craniofacial abnormalities and cutis aplasia. The structural analyses demonstrate that missense substitutions act through a dominant negative mechanism by disrupting the higher order structure of the KCTD15 protein complex.
Preoperative computed tomography-guided transscapular sens-cure needle localization for pulmonary nodule located behind the scapula.
Video-assisted thoracoscopic surgery (VATS) is an approach that is commonly used to resect pulmonary nodules (PNs). However, when these PNs are located behind the scapula, a transscapular access approach is generally required. In this study, the safety, efficacy, and feasibility of preoperative computed tomography (CT)-guided Sens-cure needle (SCN) localization was assessed for PNs located behind the scapula. From January 2020 - June 2022, a total of 122 PN patients in our hospital underwent preoperative CT-guided SCN localization and subsequent VATS resection, of whom 12 (9.8%) exhibited PNs behind the scapula necessitating a transscapular approach for this localization procedure. This study included 12 patients, each of whom had one PN located behind the scapula. The CT-guided transscapular SCN localization approach was successful in all patients, and no complications near the operative site were observed. The median localization time was 12 min, and 2 (16.7%) and 1 (8.3%) patients respectively developed pneumothorax and pulmonary hemorrhage after the localization procedure was complete. Wedge resection procedures for these PNs achieved technical success in all cases. Four patients were diagnosed with invasive adenocarcinomas and subsequently accepted lobectomy and systematic lymph node dissection. The median VATS duration and the median blood loss was 80 min and 10 mL, respectively. In total, 3, 5, and 4 PNs were respectively diagnosed as benign, mini-invasive adenocarcinomas, and invasive adenocarcinomas. Preoperative CT-guided transscapular SCN localization represents a safe, straightforward, and effective means of localizing PNs present behind the scapula.
Publicações recentes
Scalp-Ear-Nipple Syndrome: A Rare Sporadic Presentation.
A BTB extension and ion-binding domain contribute to the pentameric structure and TFAP2A binding of KCTD1.
Structural studies of KCTD1 and its disease-causing mutant P20S provide insights into the protein function and misfunction.
BTB domain mutations perturbing KCTD15 oligomerisation cause a distinctive frontonasal dysplasia syndrome.
KCTD1 and Scalp-Ear-Nipple ('Finlay-Marks') syndrome may be associated with myopia and Thin basement membrane nephropathy through an effect on the collagen IV α3 and α4 chains.
📚 EuropePMC10 artigos no totalmostrando 12
Scalp-Ear-Nipple Syndrome: A Rare Sporadic Presentation.
International journal of dermatologyA BTB extension and ion-binding domain contribute to the pentameric structure and TFAP2A binding of KCTD1.
Structure (London, England : 1993)Structural studies of KCTD1 and its disease-causing mutant P20S provide insights into the protein function and misfunction.
International journal of biological macromoleculesBTB domain mutations perturbing KCTD15 oligomerisation cause a distinctive frontonasal dysplasia syndrome.
Journal of medical geneticsPreoperative computed tomography-guided transscapular sens-cure needle localization for pulmonary nodule located behind the scapula.
Journal of cardiothoracic surgeryKCTD1 and Scalp-Ear-Nipple ('Finlay-Marks') syndrome may be associated with myopia and Thin basement membrane nephropathy through an effect on the collagen IV α3 and α4 chains.
Ophthalmic geneticsPoint Shear Wave Elastography by ElastPQ for Fibrosis Screening in Patients with NAFLD: A Prospective, Multicenter Comparison to Vibration-Controlled Elastography.
Ultraschall in der Medizin (Stuttgart, Germany : 1980)Scalp-Ear-Nipple syndrome: first report of a Potassium channel tetramerization domain-containing 1 in-frame insertion and review of the literature.
Clinical dysmorphologyBreast reconstruction via fat grafting for a patient with bilateral congenital amastia (Finlay-Marks syndrome).
Congenital anomaliesKCTD1 mutants in scalp‑ear‑nipple syndrome and AP‑2α P59A in Char syndrome reciprocally abrogate their interactions, but can regulate Wnt/β‑catenin signaling.
Molecular medicine reportsAP-2β/KCTD1 Control Distal Nephron Differentiation and Protect against Renal Fibrosis.
Developmental cellMolecular basis of the scalp-ear-nipple syndrome unraveled by the characterization of disease-causing KCTD1 mutants.
Scientific reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome couro cabeludo-pavilhão auricular-mamilo.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome couro cabeludo-pavilhão auricular-mamilo
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Scalp-Ear-Nipple Syndrome: A Rare Sporadic Presentation.
- A BTB extension and ion-binding domain contribute to the pentameric structure and TFAP2A binding of KCTD1.
- Structural studies of KCTD1 and its disease-causing mutant P20S provide insights into the protein function and misfunction.
- BTB domain mutations perturbing KCTD15 oligomerisation cause a distinctive frontonasal dysplasia syndrome.
- Preoperative computed tomography-guided transscapular sens-cure needle localization for pulmonary nodule located behind the scapula.
- KCTD1 and Scalp-Ear-Nipple ('Finlay-Marks') syndrome may be associated with myopia and Thin basement membrane nephropathy through an effect on the collagen IV α3 and α4 chains.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2036(Orphanet)
- OMIM OMIM:181270(OMIM)
- MONDO:0008404(MONDO)
- GARD:159(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7429841(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome couro cabeludo-pavilhão auricular-mamilo
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata