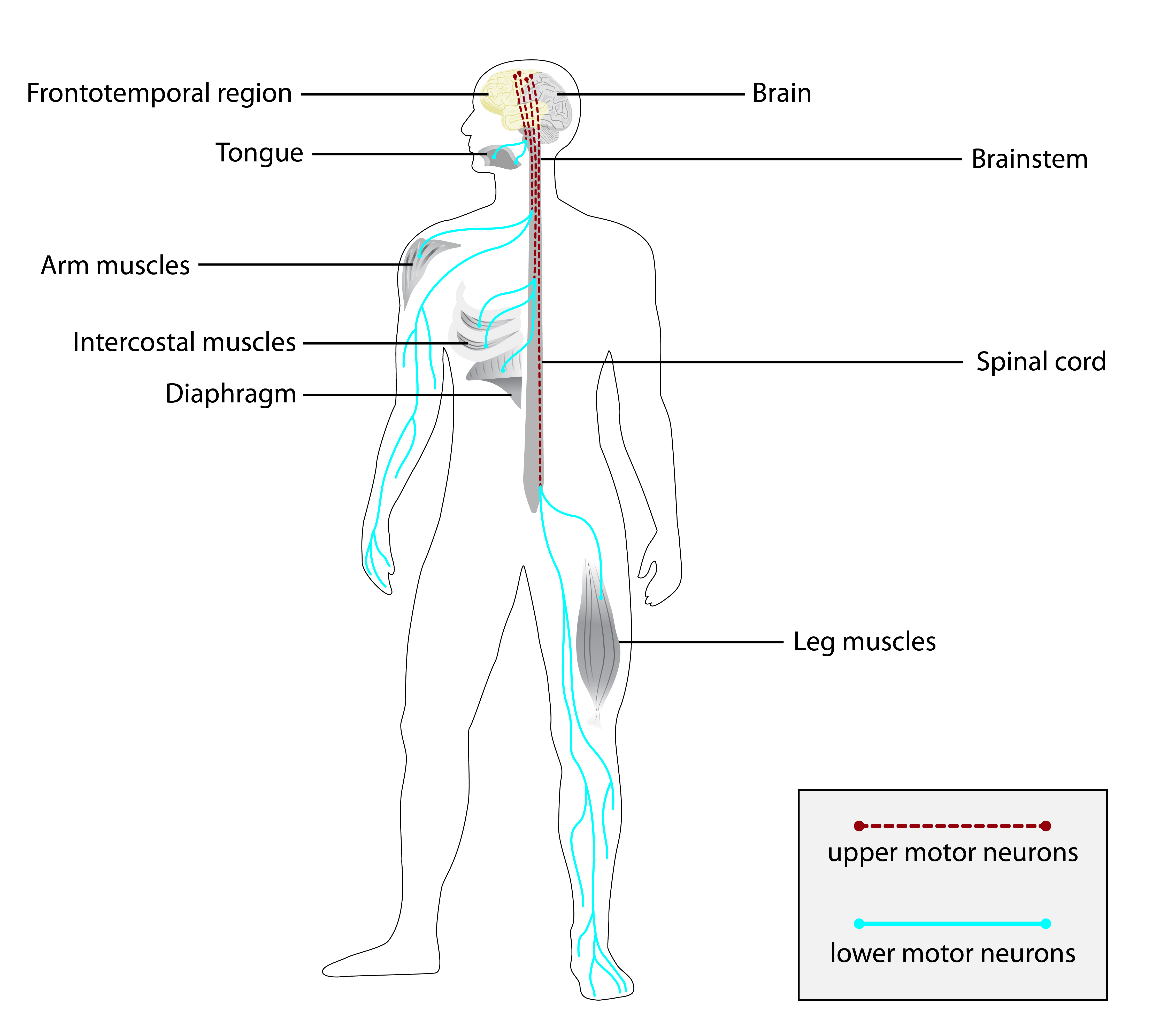
A síndrome de ataxia espástica, epilepsia mioclônica e neuropatia de início precoce é uma doença rara e hereditária (passada de pais para filhos) que causa falta de coordenação com rigidez. Ela se manifesta na infância com uma fraqueza progressiva e rigidez nas pernas, além de problemas de coordenação que pioram lentamente, incluindo dificuldade para falar, para engolir e uma perda gradual dos movimentos. Além disso, a síndrome está ligada a problemas nos nervos que afetam tanto a sensibilidade quanto os movimentos (neuropatia sensório-motora), resultando em fraqueza e perda de massa muscular nas extremidades das pernas (como pés e panturrilhas). Também causa uma forma de epilepsia que piora com o tempo, caracterizada por espasmos musculares repentinos e involuntários (mioclonias). Outros sintomas que podem aparecer incluem: problemas nos olhos (como pálpebra caída e dificuldade em mover os olhos de forma coordenada), dificuldade em controlar a precisão dos movimentos, dificuldade em fazer movimentos rápidos e repetidos, movimentos involuntários de torção ou posturas anormais (distonia) e espasmos musculares súbitos.
Introdução
O que você precisa saber de cara
A síndrome de ataxia espástica, epilepsia mioclônica e neuropatia de início precoce é uma doença rara e hereditária (passada de pais para filhos) que causa falta de coordenação com rigidez. Ela se manifesta na infância com uma fraqueza progressiva e rigidez nas pernas, além de problemas de coordenação que pioram lentamente, incluindo dificuldade para falar, para engolir e uma perda gradual dos movimentos. Além disso, a síndrome está ligada a problemas nos nervos que afetam tanto a sensibilidade quanto os movimentos (neuropatia sensório-motora), resultando em fraqueza e perda de massa muscular nas extremidades das pernas (como pés e panturrilhas). Também causa uma forma de epilepsia que piora com o tempo, caracterizada por espasmos musculares repentinos e involuntários (mioclonias). Outros sintomas que podem aparecer incluem: problemas nos olhos (como pálpebra caída e dificuldade em mover os olhos de forma coordenada), dificuldade em controlar a precisão dos movimentos, dificuldade em fazer movimentos rápidos e repetidos, movimentos involuntários de torção ou posturas anormais (distonia) e espasmos musculares súbitos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 10 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 33 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCatalytic component of the m-AAA protease, a protease that plays a key role in proteostasis of inner mitochondrial membrane proteins, and which is essential for axonal and neuron development (PubMed:19748354, PubMed:28396416, PubMed:29932645, PubMed:30683687, PubMed:31327635, PubMed:37917749, PubMed:38157846). AFG3L2 possesses both ATPase and protease activities: the ATPase activity is required to unfold substrates, threading them into the internal proteolytic cavity for hydrolysis into small pe
Mitochondrion inner membrane
Spinocerebellar ataxia 28
Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA28 is an autosomal dominant cerebellar ataxia (ADCA) with a slow progressive course and no evidence of sensory involvement or cognitive impairment.
Variantes genéticas (ClinVar)
270 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de ataxia espástica-epilepsia mioclônica-neuropatia de início precoce
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Expanding the Genetic Landscape of ATXN2 Variants: Insights From a Biallelic Trinucleotide Repeat Expansion in an Acadian Family.
Spinocerebellar ataxias are a diverse group of autosomal dominant cerebellar ataxias. SCA2 is a complex ataxia with various extracerebellar symptoms, including parkinsonism, dystonia, hyporeflexia, and cognitive impairment. ATXN2 is a modulator of neurologic disease: Expansions of at least 37 CAG (glutamine) repeats are pathogenic for SCA2, while expansions in the intermediate range (30-32) convey risk for the development of neurodegenerative disorders including Parkinson disease and amyotrophic lateral sclerosis. Homozygous variants are exceedingly rare. This study describes a novel ATXN2 presentation identified in an Acadian family from New Brunswick, Canada: a CAG repeat expansion within the fully penetrant range of SCA2, asymptomatic in the heterozygous state and resulting in a neurodegenerative disorder in homozygous patients. Three individuals, 2 siblings and their cousin, were investigated for a neurodegenerative disorder with overlapping phenotypes. The affected individuals and their 5 immediate family members underwent whole-genome sequencing analyzed by ExpansionHunter, repeat-primed PCR and Sanger sequencing. Sequencing revealed a homozygous 39/39 CAG repeat expansion with 4 CAA interruptions in ATXN2 across all 3 affected individuals. After experiencing childhood intellectual or learning difficulties, the patients developed a pyramidal syndrome with spastic gait and a major neurocognitive disorder characterized by prominent frontal signs during their late twenties. Within a decade, all patients completely lost their autonomy. Shared phenotypic features included ataxia, spasticity, aphasia, dysphagia, myoclonus, atypical parkinsonism, incontinence, diffuse cortical atrophy with frontal predominance, and cerebellar atrophy. The same 39 CAG repeat allele with 4 CAA interruptions was identified in heterozygous state in 4 asymptomatic parents (age 65+) and 1 sibling in their thirties. Three carriers consented to further investigation with a neurologic examination, neuropsychological assessment, and cerebral MRI. Clinical and radiologic signs of disease were absent, despite the carriers' ages and their heterozygous expansion in the fully penetrant range of SCA2. This study describes a novel ATXN2 expansion within the classic pathogenic range for SCA2 that manifests as an early-onset neurodegenerative disorder in the homozygous state, while being asymptomatic into late adulthood in the heterozygous state.
Novel KIF5A variant in a patient with early-onset levodopa-responsive Parkinson's syndrome.
We present the case of a male in his mid-30s with a progressive complex neurological phenotype primarily characterised by levodopa-responsive parkinsonism with motor fluctuations as well as gait ataxia, peripheral neuropathy and finally also spastic paraplegia. Genetic analysis identified a novel heterozygous variant in the KIF5A gene: c.937G>A (p.Glu313Lys). This variant is genetically classified as likely pathogenic. Other pathogenic mutations in the KIF5A gene are associated with hereditary spastic paraplegia type 10, Charcot-Marie-Tooth disease type 2 and amyotrophic lateral sclerosis. We discuss the clinical, genetic and prognostic implications of this finding.
Expanding the clinical and immunological phenotypes of COPB1 deficiency.
COPB1 encodes the coatomer subunit beta protein, which is essential for brain development and intracellular protein trafficking. Homozygous mutations cause Baralle-Macken syndrome that characterized by global developmental delay, severe intellectual disability, and early-onset cataracts. Although immunodeficiency has been observed in patients with COPB1 deficiency, the immunological phenotype remains incompletely characterized. Here, we comprehensively describe the clinical features and delineate the immunological phenotype associated with COPB1 mutations. We performed detailed clinical and immunological evaluations of three female siblings with COPB1 deficiency. Flow cytometry was used to characterize lymphocyte subsets and to assess cytokine secretion following stimulation. Functional proliferation of peripheral blood mononuclear cells (PBMCs) was assessed using dye labeling, CD3/CD28 activation, and flow cytometric analysis. Three female siblings with COPB1 deficiency presented with early-onset cataracts, global developmental delay, hypotonia, and progressive spasticity leading to quadriplegia. All patients experienced recurrent infections beginning in early childhood. Immunological evaluation revealed neutropenia, T cell lymphopenia, profound reduction in switched and unswitched memory B cells, and absent specific antibody responses. All the three patients were initiated on immunoglobulin replacement therapy and antimicrobial prophylaxis. Our findings expand the clinical and immunological spectrum of COPB1 deficiency, demonstrating combined immunodeficiency with neutropenia, lymphopenia and impaired specific antibody responses. These results support the classification of COPB1 deficiency as a combined immunodeficiency with syndromic features under the IUIS classification system and emphasize the importance of comprehensive immunological evaluation and early immunoglobulin replacement therapy in patients with COPB1 mutations.
The genetics of autosomal recessive ALS: a review of the common forms and their phenotypes.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease marked by progressive degeneration of upper and lower motor neurons. Most forms of ALS associated with a suspected causal variant are inherited in an autosomal dominant manner. However, there is an important subset of autosomal recessive (AR) variants, often associated with early-onset or atypical clinical features. Advances in genetic sequencing have led to increased recognition of AR ALS. In this review, we focus on four key confirmed AR ALS-associated genes, which appear to be most common-ALS2, SPG11, OPTN, and the D90A variant of SOD1-reviewing their pathophysiology and unique clinical manifestations. We also highlight very rare AR mutations implicated in ALS, including SYNE1, ATP13A2, and FUS, and some associated with overlap syndromes or debated pathogenicity including SIGMAR1, ERLIN1, and ERLIN2. These genes are involved in an array of processes including axonal transport, endosomal trafficking, oxidative stress response, and autophagy, suggesting distinct mechanisms of motor neuron degeneration. Some forms of AR ALS more frequently present with juvenile onset and slower progression, but other genes are associated with broader phenotypic spectra. This includes overlap with hereditary spastic paraplegia (HSP) and hereditary ataxias. Understanding these AR forms of ALS may enhance diagnostic precision, improve prognostication, and may pave the way for targeted gene therapies. This review underscores the emerging significance of AR inheritance in ALS and calls for deeper investigation into its molecular and clinical dimensions.
The Age of Definitive Fusion Surgery for Early Onset Scoliosis Has Remained Constant Over the Past 2 Decades.
Treatment options for early-onset scoliosis (EOS) are confounded by the risks associated with intervention at a younger age. Spinal instrumentation must be considered carefully due to potentially adverse effects to the spine, chest wall, and lungs. Posterior spinal fusion before subsequent growth can also lead to the crankshaft phenomenon. With the recent increasing interest in delaying spinal fusion, we aim to determine trends in patient age selection at a single definitive (termed "one and done") fusion for EOS. We identified 791 patients from 2005 to 2022 who met the inclusion criteria (age 5 y or younger, single definitive fusion, and complete data). Patients who underwent a hemivertebrae resection with limited fusion were not included. Multiple linear regression was performed with date of fusion as the independent variable and age at definitive fusion as the dependent variable. Our regression included race and sex to control for their effects as confounders. We repeated this analysis with groups separated by scoliosis etiology and sex. Coefficients with P<0.05 were considered significant. In the entire cohort, there was no significant change in the age at definitive fusion between 2005 and 2022 (coefficient=0.042, P=0.099). The mean age at fusion was 12.1 years. Of these, 167 (21.1%) cases had congenital scoliosis, 277 (35.0%) had idiopathic scoliosis, 191 (24.1%) had neuromuscular scoliosis, and 156 (19.7%) had syndromic scoliosis. Patients with idiopathic (-0.002, P=0.962), syndromic (-0.027, P=0.671), and neuromuscular (-0.005, P=0.924) EOS showed no significant change in the age at fusion. However, children with congenital EOS (0.171, P=0.006) and females (0.082, P=0.003) demonstrated a significant increase. On the basis of our regression models, the predicted age at definitive fusion increased from 10.6 years to 12.3 years in those with congenital EOS and from 11.4 to 12.1 years in females. Over a 17-year study period, females and congenital EOS patients demonstrated significant increases in age at the time of definite fusion. There was no significant change for children with neuromuscular, idiopathic, or syndromic EOS over the same time frame. Further study is necessary to determine the nature of these disparities.
Publicações recentes
[A case of rare hereditary Siddiqi syndrome with novel neuropsychiatric signs].
New Insights Into TRMT10A Syndrome: Case Report and Literature Review.
TCEAL1 loss-of-function results in an X-linked dominant neurodevelopmental syndrome and drives the neurological disease trait in Xq22.2 deletions.
De Novo ATP1A1 Variants in an Early-Onset Complex Neurodevelopmental Syndrome.
[Resistant epileptic encephalopathy in a child with microcephalic capillary malformation syndrome].
📚 EuropePMCmostrando 110
Expanding the clinical and immunological phenotypes of COPB1 deficiency.
Frontiers in immunologyExpanding the Genetic Landscape of ATXN2 Variants: Insights From a Biallelic Trinucleotide Repeat Expansion in an Acadian Family.
Neurology. GeneticsNovel KIF5A variant in a patient with early-onset levodopa-responsive Parkinson's syndrome.
BMJ case reportsThe genetics of autosomal recessive ALS: a review of the common forms and their phenotypes.
Amyotrophic lateral sclerosis & frontotemporal degenerationThe Age of Definitive Fusion Surgery for Early Onset Scoliosis Has Remained Constant Over the Past 2 Decades.
Journal of pediatric orthopedicsSpinocerebellar ataxias masquerading as movement disorders: clinical and genetic characterization.
Frontiers in neurologyKohlschütter-Tönz Syndrome: A Rare Clinical Entity with Amelogenesis Imperfecta in Two Siblings, Dental Management and Scoping Review.
Turkish archives of pediatricsClinical and genetic analysis of ERCC8-Related cockayne syndrome: hepatic dysfunction as a biomarker, anhidrosis as a rare feature, and rehabilitation outcomes for ankle contractures.
Frontiers in geneticsCase Report: Novel ATP13A2 pathogenic variants associated with early-onset parkinsonism and a mini-review.
Frontiers in geneticsFrench protocol for diagnosis and management of type 1 interferonopathies.
La Revue de medecine interneA Novel TAF1C Missense Variant Causes Neurodevelopmental Regression via Disrupted Nucleolar Localization and Nucleoplasmic Aggregation.
Clinical genetics[A case of rare hereditary Siddiqi syndrome with novel neuropsychiatric signs].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaThe phenotypic spectrum of PTCD3 deficiency.
JIMD reportsNew Insights Into TRMT10A Syndrome: Case Report and Literature Review.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsPhenotypic variability related to dominant UCHL1 mutations: about three families with optic atrophy and ataxia.
Journal of neurologyAdult-onset deletion of ATP13A2 in mice induces progressive nigrostriatal pathway dopaminergic degeneration and lysosomal abnormalities.
NPJ Parkinson's diseaseCase report: Early-onset Parkinson's disease with lower limb spasticity in a new DJ-1/PARK7 patient.
Frontiers in neuroscienceHereditary spastic paraparesis type 18 (SPG18): new ERLIN2 variants in a series of Italian patients, shedding light upon genetic and phenotypic variability.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyType 1 early infantile epileptic encephalopathy: A case report and literature review.
Molecular genetics & genomic medicineHereditary spastic paraparesis type 46 (SPG46): new GBA2 variants in a large Italian case series and review of the literature.
NeurogeneticsATP13A2 (PARK9) and basal ganglia function.
Frontiers in neurologyGeneration of three induced pluripotent stem cell lines from individuals with Aicardi-Goutières syndrome caused by a c.3019G>A (p.G1007R) autosomal dominant pathogenic variant in ADAR1.
Stem cell researchThe Rogdi knockout mouse is a model for Kohlschütter-Tönz syndrome.
Scientific reportsVestibular Hypofunction in ARSACS Syndrome: A Possible Pitfall in the Differential Diagnosis of Recessive Cerebellar and Afferent Ataxias.
Neurology. Clinical practiceGRM7-related disorder: five additional patients from three independent families and review of the literature.
European journal of medical geneticsBi-allelic ACBD6 variants lead to a neurodevelopmental syndrome with progressive and complex movement disorders.
Brain : a journal of neurologyJuvenile Dermatomyositis and Infantile Cerebral Palsy: Aicardi-Gouteres Syndrome, Type 5, with a Novel Mutation in SAMHD1-A Case Report.
BiomedicinesCase report: Mohr-Tranebjaerg syndrome: hearing impairment as the onset of an insidious disorder with high recurrence risk.
Frontiers in neurologyRNASEH2C c.194G>A is a Chinese-specific founder mutation in three unrelated patients with Aicardi-Goutières syndrome 3.
Clinical geneticsNovel SERAC1 Variant Presenting With Adult-Onset Extrapyramidal Dystonia-Parkinsonism Phenotype: A Case Report.
Neurology. GeneticsThe clinical and genetic spectrum of autosomal-recessive TOR1A-related disorders.
Brain : a journal of neurologyRapidly Progressive Frontotemporal Dementia With Amyotrophic Lateral Sclerosis in an Elderly Female.
CureusCase report: Early-onset Parkinson's disease with initial spastic paraparesis and hyperreflexia caused by compound heterozygous PRKN-gene exon 2 and 4 deletions.
Frontiers in neurologyTCEAL1 loss-of-function results in an X-linked dominant neurodevelopmental syndrome and drives the neurological disease trait in Xq22.2 deletions.
American journal of human geneticsExpanding SPG7 dominant optic atrophy phenotype: Infantile nystagmus and optic atrophy without spastic paraplegia.
American journal of medical genetics. Part APhenotypic continuum of NFU1-related disorders.
Annals of clinical and translational neurologyA Novel de novo KIF1A Mutation in a Patient with Ataxia, Intellectual Disability and Mild Foot Deformity.
Cerebellum (London, England)Early-Onset and Severe Complex Hereditary Spastic Paraplegia Caused by De Novo Variants in SPAST.
Movement disorders : official journal of the Movement Disorder SocietyBi-allelic loss-of-function variants in PPFIBP1 cause a neurodevelopmental disorder with microcephaly, epilepsy, and periventricular calcifications.
American journal of human geneticsOne Molecule for Mental Nourishment and More: Glucose Transporter Type 1-Biology and Deficiency Syndrome.
BiomedicinesAdult-Onset Neurodegeneration in Nucleotide Excision Repair Disorders (NERDND ): Time to Move Beyond the Skin.
Movement disorders : official journal of the Movement Disorder SocietyEl-Hattab-Alkuraya syndrome caused by biallelic WDR45B pathogenic variants: Further delineation of the phenotype and genotype.
Clinical geneticsNovel CAPN1 missense variants in complex hereditary spastic paraplegia with early-onset psychosis.
Annals of clinical and translational neurologyNovel SEPSECS Pathogenic Variants Featuring Unusual Phenotype of Complex Movement Disorder With Thin Corpus Callosum: A Case Report.
Neurology. GeneticsMotor and behavioral phenotype of Dravet syndrome in adulthood.
Epilepsy & behavior : E&BDe Novo ATP1A1 Variants in an Early-Onset Complex Neurodevelopmental Syndrome.
NeurologyMultifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature.
Frontiers in neurologySPG6 (NIPA1 variant): A report of a case with early-onset complex hereditary spastic paraplegia and brief literature review.
Journal of clinical neuroscience : official journal of the Neurosurgical Society of AustralasiaVery Early-Onset Alzheimer's Disease in the Third Decade of Life with de novo PSEN1 Mutations.
Journal of Alzheimer's disease : JADPresenilin-1 Mutations Are a Cause of Primary Lateral Sclerosis-Like Syndrome.
Frontiers in molecular neuroscienceDe Novo PS1 Mutation (Pro436Gln) in a Very Early-Onset Posterior Variant of Alzheimer's Disease Associated with Spasticity: A Case Report.
Journal of Alzheimer's disease : JADThe spectrum of neurodevelopmental, neuromuscular and neurodegenerative disorders due to defective autophagy.
AutophagyA Novel Approach to New-Onset Hemiplegic Shoulder Pain With Decreased Range of Motion Using Targeted Diagnostic Nerve Blocks: The ViVe Algorithm.
Frontiers in neurologyNGS-Based Diagnosis of Treatable Neurogenetic Disorders in Adults: Opportunities and Challenges.
GenesNeuro-Ophthalmic Phenotype of OPA3.
Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology SocietyBlended Phenotype of Silver-Russell Syndrome and SPG50 Caused by Maternal Isodisomy of Chromosome 7.
Neurology. GeneticsDefining the clinical, molecular and imaging spectrum of adaptor protein complex 4-associated hereditary spastic paraplegia.
Brain : a journal of neurology[Resistant epileptic encephalopathy in a child with microcephalic capillary malformation syndrome].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaA relatively common homozygous TRAPPC4 splicing variant is associated with an early-infantile neurodegenerative syndrome.
European journal of human genetics : EJHGHeterozygous KIF1A variants underlie a wide spectrum of neurodevelopmental and neurodegenerative disorders.
Journal of medical geneticsNovel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report.
GenesMonogenic lupus due to spondyloenchondrodysplasia with spastic paraparesis and intracranial calcification: case-based review.
Rheumatology internationalLeigh syndrome in a patient with a novel C12orf65 pathogenic variant: case report and literature review.
Genetics and molecular biologySpinocerebellar ataxia type 48: last but not least.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyDiagnosis of Aicardi-Goutières Syndrome in Adults: A Case Series.
Movement disorders clinical practiceClinical features of inherited neuropathy with BSCL2 mutations in Japan.
Journal of the peripheral nervous system : JPNSNovel mutations in the KCNJ10 gene associated to a distinctive ataxia, sensorineural hearing loss and spasticity clinical phenotype.
NeurogeneticsA VPS13D spastic ataxia mutation disrupts the conserved adaptor-binding site in yeast Vps13.
Human molecular geneticsDeficiencies in vesicular transport mediated by TRAPPC4 are associated with severe syndromic intellectual disability.
Brain : a journal of neurologyCompound heterozygous mutations in SNAP29 is associated with Pelizaeus-Merzbacher-like disorder (PMLD).
Human geneticsXq22 deletions and correlation with distinct neurological disease traits in females: Further evidence for a contiguous gene syndrome.
Human mutationCharcot-Marie-Tooth disease and related disorders: an evolving landscape.
Current opinion in neurologyDefining the clinical-genetic and neuroradiological features in SPG54: description of eight additional cases and nine novel DDHD2 variants.
Journal of neurologyAxonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome.
AutophagyGlucose Transporter Type 1 Deficiency Syndrome: Developmental Delay and Early-Onset Ataxia in a Novel Mutation of the SLC2A1 Gene.
Journal of pediatric neurosciences[Proteins from Vps13 family: from molecular function to pathogenesis of neurodegenerative disorders].
Postepy biochemiiMutations in ACTL6B, coding for a subunit of the neuron-specific chromatin remodeling complex nBAF, cause early onset severe developmental and epileptic encephalopathy with brain hypomyelination and cerebellar atrophy.
Human geneticsElectroencephalographic and epilepsy findings in mecp2 duplication syndrome. A family study.
Brain & developmentWest syndrome, developmental and epileptic encephalopathy, and severe CNS disorder associated with WWOX mutations.
Epileptic disorders : international epilepsy journal with videotapeConcurrent AFG3L2 and SPG7 mutations associated with syndromic parkinsonism and optic atrophy with aberrant OPA1 processing and mitochondrial network fragmentation.
Human mutationThe glucose transporter type 1 (Glut1) syndromes.
Epilepsy & behavior : E&BDiseases of ganglioside biosynthesis: An expanding group of congenital disorders of glycosylation.
Molecular genetics and metabolism[Hereditary optic neuropathies in pediatric ophthalmology].
Journal francais d'ophtalmologieKARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature.
Orphanet journal of rare diseasesNFU1 -Related Disorders as Key Differential Diagnosis of Cavitating Leukoencephalopathy.
Journal of pediatric geneticsDeep brain stimulation for dystonia due to cerebral palsy: A review.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyBiallelic Variants in CNPY3, Encoding an Endoplasmic Reticulum Chaperone, Cause Early-Onset Epileptic Encephalopathy.
American journal of human geneticsDifferent Cerebellar Ataxia Phenotypes Associated with Mutations of the PNPLA6 Gene in Brazilian Patients with Recessive Ataxias.
Cerebellum (London, England)Amino acid synthesis deficiencies.
Journal of inherited metabolic diseaseThe novel PSEN1 M84V mutation associated to frontal dysexecutive syndrome, spastic paraparesis, and cerebellar atrophy in a dominant Alzheimer's disease family.
Neurobiology of agingWDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: A consistent neurodevelopmental syndrome.
Clinical geneticsNot only dominant, not only optic atrophy: expanding the clinical spectrum associated with OPA1 mutations.
Orphanet journal of rare diseasesHypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia.
Brain : a journal of neurologySTUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations.
Orphanet journal of rare diseasesAutosomal-Recessive Mutations in AP3B2, Adaptor-Related Protein Complex 3 Beta 2 Subunit, Cause an Early-Onset Epileptic Encephalopathy with Optic Atrophy.
American journal of human geneticsInfantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: An update for the diagnosis.
Brain & developmentNeurodegeneration With Brain Iron Accumulation (NBIA) Syndromes Presenting With Late-Onset Craniocervical Dystonia: An Illustrative Case Series.
Movement disorders clinical practiceDelayed Induction of Human NTE (PNPLA6) Rescues Neurodegeneration and Mobility Defects of Drosophila swiss cheese (sws) Mutants.
PloS oneEarly-onset spastic paraparesis as presenting sign of familial Creutzfeldt-Jakob disease.
Parkinsonism & related disordersSevere CNS involvement in WWOX mutations: Description of five new cases.
American journal of medical genetics. Part AThe Effects of Ketogenic Diet on Seizures, Cognitive Functions, and Other Neurological Disorders in Classical Phenotype of Glucose Transporter 1 Deficiency Syndrome.
NeuropediatricsAtypical presentation of Costeff syndrome-severe psychomotor involvement and electrical status epilepticus during slow wave sleep.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyDo Glut1 (glucose transporter type 1) defects exist in epilepsy patients responding to a ketogenic diet?
Epilepsy researchContiguous mutation syndrome in the era of high-throughput sequencing.
Molecular genetics & genomic medicineHomozygous p.V116* mutation in C12orf65 results in Leigh syndrome.
Journal of neurology, neurosurgery, and psychiatryExome analysis identified a novel missense mutation in the CLPP gene in a consanguineous Saudi family expanding the clinical spectrum of Perrault Syndrome type-3.
Journal of the neurological sciencesBrief Report: IFIH1 Mutation Causes Systemic Lupus Erythematosus With Selective IgA Deficiency.
Arthritis & rheumatology (Hoboken, N.J.)Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia.
Movement disorders : official journal of the Movement Disorder SocietyThe neuropsychological profile of patients with 3-methylglutaconic aciduria type III, Costeff syndrome.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsSPTAN1 encephalopathy: distinct phenotypes and genotypes.
Journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de ataxia espástica-epilepsia mioclônica-neuropatia de início precoce.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de ataxia espástica-epilepsia mioclônica-neuropatia de início precoce
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Expanding the Genetic Landscape of ATXN2 Variants: Insights From a Biallelic Trinucleotide Repeat Expansion in an Acadian Family.
- Novel KIF5A variant in a patient with early-onset levodopa-responsive Parkinson's syndrome.
- Expanding the clinical and immunological phenotypes of COPB1 deficiency.
- The genetics of autosomal recessive ALS: a review of the common forms and their phenotypes.
- The Age of Definitive Fusion Surgery for Early Onset Scoliosis Has Remained Constant Over the Past 2 Decades.
- [A case of rare hereditary Siddiqi syndrome with novel neuropsychiatric signs].
- New Insights Into TRMT10A Syndrome: Case Report and Literature Review.
- TCEAL1 loss-of-function results in an X-linked dominant neurodevelopmental syndrome and drives the neurological disease trait in Xq22.2 deletions.
- De Novo ATP1A1 Variants in an Early-Onset Complex Neurodevelopmental Syndrome.
- [Resistant epileptic encephalopathy in a child with microcephalic capillary malformation syndrome].
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:313772(Orphanet)
- OMIM OMIM:614487(OMIM)
- MONDO:0013776(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:17409(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21097760(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de ataxia espástica-epilepsia mioclônica-neuropatia de início precoce
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub