A Síndrome da Deficiência de Creatina (SDC) é um grupo de condições genéticas, ou seja, problemas que já nascem com a pessoa, e que afetam a forma como o corpo processa a creatina. Ela se manifesta com um atraso geral no desenvolvimento, deficiência intelectual e outros sintomas neurológicos, como convulsões, dificuldades de movimento e fraqueza muscular, além de problemas de comportamento. A SDC engloba dois tipos de problemas na produção de creatina: a deficiência da enzima guanidinoacetato metiltransferase e a deficiência da enzima L-Arginina:glicina amidinotransferase. Inclui também a deficiência do transportador de creatina ligada ao cromossomo X.
Introdução
O que você precisa saber de cara
A Síndrome da Deficiência de Creatina (SDC) é um grupo de condições genéticas, ou seja, problemas que já nascem com a pessoa, e que afetam a forma como o corpo processa a creatina. Ela se manifesta com um atraso geral no desenvolvimento, deficiência intelectual e outros sintomas neurológicos, como convulsões, dificuldades de movimento e fraqueza muscular, além de problemas de comportamento. A SDC engloba dois tipos de problemas na produção de creatina: a deficiência da enzima guanidinoacetato metiltransferase e a deficiência da enzima L-Arginina:glicina amidinotransferase. Inclui também a deficiência do transportador de creatina ligada ao cromossomo X.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 45 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 102 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive, Not applicable, X-linked recessive.
Transamidinase that catalyzes the transfer of the amidino group of L-arginine onto the amino moiety of acceptor metabolites such as glycine, beta-alanine, gamma-aminobutyric acid (GABA) and taurine yielding the corresponding guanidine derivatives (PubMed:16820567, PubMed:27233232, PubMed:36543883, PubMed:3800397). Catalyzes the rate-limiting step of creatine biosynthesis, namely the transfer of the amidino group from L-arginine to glycine to generate guanidinoacetate, which is then methylated by
Mitochondrion inner membraneCytoplasm
Cerebral creatine deficiency syndrome 3
An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life.
Creatine:sodium symporter which mediates the uptake of creatine (PubMed:17465020, PubMed:22644605, PubMed:25861866, PubMed:7945388, PubMed:7953292, PubMed:9882430). Plays an important role in supplying creatine to the brain via the blood-brain barrier (By similarity)
Cell membraneApical cell membrane
Cerebral creatine deficiency syndrome 1
An X-linked disorder of creatine transport characterized by intellectual disability, severe speech delay, behavioral abnormalities, and seizures. Carrier females may show mild neuropsychologic impairment.
Converts guanidinoacetate to creatine, using S-adenosylmethionine as the methyl donor (PubMed:24415674, PubMed:26003046, PubMed:26319512). Important in nervous system development (PubMed:24415674)
Cerebral creatine deficiency syndrome 2
An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids.
Variantes genéticas (ClinVar)
894 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 622 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de deficiência de creatina
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
2 ensaios clínicos encontrados.
Publicações mais relevantes
Creatine Deficiency Syndrome: Significance of Magnetic Resonance Spectroscopy.
Structural insights into the substrate uptake and inhibition of the human creatine transporter (hCRT).
Creatine plays a vital role in cellular energy production and adenosine triphosphate (ATP) homeostasis and has also been identified as a neurotransmitter in the mammalian brain. Creatine is transported into cells by the human creatine transporter (hCRT) (SLC6A8), an Na+/Cl--dependent symporter encoded on the X chromosome. Mutations in hCRT cause cerebral creatine deficiency syndrome 1, a neurological disorder marked by intellectual disability, speech delay, and seizures. Beyond its role in the brain and muscle, hCRT is highly expressed in metabolically active tumors. Many cancer cells, including colorectal cancer and glioblastoma, upregulate hCRT to sustain intracellular creatine levels and buffer ATP under energy stress. Pharmacological blockade of hCRT by RGX202 has been shown to impair tumor growth by disrupting energy homeostasis. Here, we report the high-resolution cryo-Electron Microscopy (cryo-EM) structures of human hCRT in three states: apo, creatine-bound, and RGX202-bound. hCRT adopts a canonical LeuT-fold with 12 transmembrane helices and two pseudosymmetric inverted repeats. Creatine is coordinated in the central substrate-binding site through interactions with transmembrane helices TM1, TM3, TM6, and TM8, while the inhibitor RGX202 occupies the same binding pocket, engaging in overlapping contacts that competitively block creatine access. Our structural and mechanistic findings clarify substrate recognition and inhibitory binding of hCRT, providing a molecular rationale for targeting hCRT in both inherited metabolic diseases and cancer therapy.
Gene delivery of AGAT and GAMT boosts creatine levels in creatine transporter deficiency patient fibroblasts.
Creatine is a critical metabolite used to buffer cellular energy demands in highly energetic tissues such as the brain and muscle. Genetic defects in endogenous creatine synthesis or transport across cellular membranes lead to a common set of phenotypes referred to as Cerebral Creatine Deficiency Syndrome (CCDS). The most common form of CCDS is Creatine Transporter 1 (CT1) Deficiency (CTD). It accounts for ~ 70% of cases and results from loss-of-function mutations in the X-linked gene SLC6A8. Affected individuals suffer from intellectual disability, autistic-like behaviors, and epilepsy. There are currently no effective therapies for this disorder, but gene therapy has emerged as a potential approach. The two enzymes which comprise the endogenous creatine synthetic pathway (AGAT and GAMT) are selectively expressed by specific cell types throughout the body. However, after synthesized, creatine uptake relies on the protein product of SLC6A8, CT1, to transport creatine into target cell types. We hypothesized that gene delivery of GATM (encoding AGAT) and GAMT into end-user cell types would bypass the need for CT1, allowing for intracellular synthesis of creatine. We tested this strategy in two human cell types: HEK293T cells and primary fibroblasts. Co-delivery of GATM and GAMT increased internal creatine concentrations by 7.6-fold in HEK293T cells and 12.3-fold in healthy control fibroblasts. We then employed this approach to primary fibroblasts from patients with CTD. This resulted in an up to 11.6-fold increase in intracellular creatine concentrations, far exceeding the intracellular concentration of creatine in healthy control fibroblasts. Importantly, overexpression of AGAT and GAMT resulted in proper targeting of these enzymes to their natural cellular compartment and did not impair the growth of patient fibroblasts. These findings establish gene therapy with GATM and GAMT as a potential strategy for patients with CTD.
[Two cases of creatine deficiency syndrome caused by GAMT gene mutations and literature review].
To summarize the clinical manifestations and genetic characteristics of creatine deficiency syndrome (CDS) caused by GAMT gene mutations. A retrospective analysis was conducted on the clinical and genetic data of two children diagnosed with GAMT deficiency-type CDS at the Children's Medical Center of Xiangya Hospital, Central South University, from December 2020 to December 2024. The two patients presented with symptoms in infancy, and both had compound heterozygous mutations in the GAMT gene. Case 1 exhibited seizures and intellectual disability, while Case 2 had intellectual disability and attention-deficit hyperactivity disorder. Magnetic resonance spectroscopy of cranial MRI in both patients indicated reduced creatine peaks. After creatine treatment, seizures in Case 1 were controlled, but both patients continued to experience intellectual disabilities and behavioral issues. As of December 2024, a total of 21 cases have been reported in China (including this study), and 115 cases have been reported abroad. All patients exhibited developmental delay or intellectual disabilities, with 66.9% (91/136) experiencing seizures, 33.8% (46/136) presenting with motor disorders, and 36.8% (50/136) having behavioral problems. Seventy-five percent (102/136) of patients received creatine treatment, leading to significant improvements in seizures and motor disorders, although cognitive improvement was not substantial. GAMT deficiency-type CDS is rare and presents with nonspecific clinical features. Timely diagnosis facilitates targeted treatment, which can partially improve prognosis. 目的: 总结GAMT基因变异所致肌酸缺乏综合征(creatine deficiency syndrome, CDS)的临床表现及遗传学特点。方法: 回顾性分析2020年12月—2024年12月中南大学湘雅医院儿童医学中心诊治的2例GAMT缺乏型CDS患儿的临床和遗传学资料。结果: 2例患儿均为婴幼儿期起病,GAMT基因均存在复合杂合变异,病例1表现为癫痫发作和智力障碍,病例2存在智力障碍及注意力缺陷多动障碍;2例患儿头颅磁共振波谱提示肌酸峰减低。肌酸治疗后病例1癫痫发作控制,2例患儿仍存在智力障碍及行为问题。截至2024年12月,国内共报道21例患者(包括本研究),国外报道115例。所有患者均有发育迟缓或智力障碍,66.9%(91/136)患者有癫痫发作,33.8%(46/136)患者存在运动障碍,36.8%(50/136)患者有行为问题,75.0%(102/136)患者接受肌酸治疗,癫痫发作及运动障碍可明显改善,认知改善不明显。结论: GAMT缺乏型CDS罕见且临床表现不特异,及时诊断有助于针对性治疗,可部分改善预后。.
Imaging the Future: Diagnosing Treatable Neurometabolic Disorders in Children.
Inborn errors of metabolism are a prevalent cause of pediatric neurological abnormalities, often resulting from enzyme deficiencies that disrupt metabolic pathways. Understanding the radiological manifestations of these disorders is critical for timely diagnosis and therapy. This study aimed to elucidate the magnetic resonance imaging (MRI) brain characteristics and magnetic resonance spectroscopy (MRS) findings of specific treatable pediatric neurometabolic disorders through clinical vignettes, thereby enhancing diagnostic accuracy and informing therapeutic approaches. The study includes four cases with varying presentations, all confirmed by genetic testing, and utilized magnetic resonance imaging and spectroscopy to identify characteristic features. The first case, diagnosed as thiamine metabolism dysfunction syndrome type 4, exhibits bilateral corpus striatum T2 hyperintensities with diffusion restriction on MRI brain, increased lactate peak, and reduced N-acetylaspartate (NAA) on magnetic resonance spectroscopy (MRS), and SLC25A19 gene mutation on genetic analysis - features consistent with biotin-thiamine-responsive basal ganglia disease. The second case, identified as thiamine metabolism dysfunction syndrome type 2, reveals T2 hyperintensities in the bilateral corpus striatum, medial thalami, and cerebellar white matter, along with restricted diffusion, reduced NAA, and an inverted lactate doublet on MRS, with genetic analysis confirming an SLC19A3 mutation, also falling under the spectrum of biotin-thiamine-responsive basal ganglia disease. The third case, representing cerebral creatine deficiency syndrome type 2, demonstrates bilateral symmetric T2 hyperintensities in the globus pallidus and central tegmental tracts, a characteristic absence of a creatine peak on MRS, and a confirmed guanidomethyl transferase (GAMT) deficiency gene mutation. The fourth case, diagnosed as hypermanganesemia with dystonia type 2, is characterized by T1 hyperintensity and T2 hypointensity involving the bilateral globus pallidi, substantia nigra, and dentate nuclei, with genetic testing revealing an SLC39A14 mutation. The study emphasizes the importance of MR imaging and spectroscopy in identifying pediatric neurometabolic diseases that can be treated.
Publicações recentes
Transporters for Creatine and Related Guanidino Compounds: Their Relevance to Brain Health and Disorders.
Creatine Deficiency Syndrome: Significance of Magnetic Resonance Spectroscopy.
Imaging the Future: Diagnosing Treatable Neurometabolic Disorders in Children.
Structural insights into the substrate uptake and inhibition of the human creatine transporter (hCRT).
Dynamic electro-clinical features in Guanidinoacetate N-methyltransferase deficiency: A familial case series.
📚 EuropePMC28 artigos no totalmostrando 47
Creatine Deficiency Syndrome: Significance of Magnetic Resonance Spectroscopy.
Indian journal of pediatricsImaging the Future: Diagnosing Treatable Neurometabolic Disorders in Children.
CureusStructural insights into the substrate uptake and inhibition of the human creatine transporter (hCRT).
Proceedings of the National Academy of Sciences of the United States of AmericaDynamic electro-clinical features in Guanidinoacetate N-methyltransferase deficiency: A familial case series.
Epilepsia openGene delivery of AGAT and GAMT boosts creatine levels in creatine transporter deficiency patient fibroblasts.
PloS one[Two cases of creatine deficiency syndrome caused by GAMT gene mutations and literature review].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsValidation and Optimization of a Stable Isotope-Labeled Substrate Assay for Measuring AGAT Activity.
International journal of molecular sciencesReduced guanidinoacetate in plasma of patients with autosomal dominant Fanconi syndrome due to heterozygous P341L GATM variant and study of organoids towards treatment.
JIMD reportsFusogenic Liposomes for the Intracellular Delivery of Phosphocreatine.
Pharmaceuticals (Basel, Switzerland)Efficacy of creatine supplementation in a patient with epilepsy with SLC6A8 gene mutations.
Epileptic disorders : international epilepsy journal with videotapeExperimental and Computational Analysis of Newly Identified Pathogenic Mutations in the Creatine Transporter SLC6A8.
Journal of molecular biology[Clinical and genetic analysis of a child with Cerebral creatine deficiency syndrome due to variant of SLC6A8 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsGuanidinoacetate N-methyltransferase deficiency: Case report and brief review of the literature.
Radiology case reportsFourteen cases of cerebral creatine deficiency syndrome in children: a cohort study in China.
Translational pediatricsNovel guanidinoacetate methyltransferase (GAMT) mutation associated with cerebral creatine deficiency syndrome in a Syrian child: a case report.
Annals of medicine and surgery (2012)Cerebral Creatine Deficiency Affects the Timing of Oligodendrocyte Myelination.
The Journal of neuroscience : the official journal of the Society for NeuroscienceExpanding the neuroimaging findings of guanidinoacetate methyltransferase deficiency in an Iranian girl with a homozygous frameshift variant in the GAMT.
NeurogeneticsInborn Errors of Metabolism Associated With Autism Among Children: A Multicenter Study from Iran.
Indian pediatricsIdentification of novel variations in SLC6A8 and GAMT genes causing cerebral creatine deficiency syndrome.
Clinica chimica acta; international journal of clinical chemistryClinical diagnosis of metabolic disorders using untargeted metabolomic profiling and disease-specific networks learned from profiling data.
Scientific reports[Clinical characterization and genetic testing for a patient with creatine deficiency syndrome 1].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsSLC gene mutations and pediatric neurological disorders: diverse clinical phenotypes in a Saudi Arabian population.
Human geneticsAutism spectrum disorder in patients with inherited metabolic disorders-a large sample from a tertiary center.
The Turkish journal of pediatricsGATM and GAMT synthesize creatine locally throughout the mammalian body and within oligodendrocytes of the brain.
Brain research[Analysis of clinical features and genetic variants in a child with creatine deficiency syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsThe Creatine Transporter Unfolded: A Knotty Premise in the Cerebral Creatine Deficiency Syndrome.
Frontiers in synaptic neuroscienceLocus heterogeneity in two siblings presenting with developmental delay, intellectual disability and autism spectrum disorder.
Gene[Genetic analysis and treatment for an infant with cerebral creatine deficiency syndrome type 2].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsGuanidinoacetate Methyltransferase (GAMT) Deficiency, A Cerebral Creatine Deficiency Syndrome: A Rare Treatable Metabolic Disorder.
Annals of Indian Academy of NeurologyCase Series of Creatine Deficiency Syndrome due to Guanidinoacetate Methyltransferase Deficiency.
Annals of Indian Academy of Neurology[Clinical features and SLC6A8 gene mutations of cerebral creatine deficiency syndrome I: an analysis of two families].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsThe phenotype-driven computational analysis yields clinical diagnosis for patients with atypical manifestations of known intellectual disability syndromes.
Molecular genetics & genomic medicineClassification of the Molecular Defects Associated with Pathogenic Variants of the SLC6A8 Creatine Transporter.
BiochemistryNeuroimaging Spectrum of Inherited Neurotransmitter Disorders.
NeuropediatricsPriorities for Newborn Screening of Genetic Epilepsy.
Pediatric neurologyPrimary creatine deficiency syndrome as a potential missed diagnosis in children with psychomotor delay and seizure: case presentation with two novel variants and literature review.
Acta neurologica BelgicaRescue by 4-phenylbutyrate of several misfolded creatine transporter-1 variants linked to the creatine transporter deficiency syndrome.
NeuropharmacologyA Nervous System-Specific Model of Creatine Transporter Deficiency Recapitulates the Cognitive Endophenotype of the Disease: a Longitudinal Study.
Scientific reportsCell-Type-Specific Spatiotemporal Expression of Creatine Biosynthetic Enzyme S-adenosylmethionine:guanidinoacetate N-methyltransferase in Developing Mouse Brain.
Neurochemical researchVariability of Creatine Metabolism Genes in Children with Autism Spectrum Disorder.
International journal of molecular sciences[MR spectroscopy in metabolic disorders of the brain].
Der RadiologeCreatine Deficiency Syndrome could be Missed Easily: A Case Report of Guanidinoacetate Methyltransferase Deficiency Presented with Neurodevelopmental Delay, Seizures, and Behavioral Changes, but Normal Structural MRI.
Annals of clinical and laboratory scienceA mouse model for creatine transporter deficiency reveals early onset cognitive impairment and neuropathology associated with brain aging.
Human molecular geneticsSystemic availability of guanidinoacetate affects GABAA receptor function and seizure threshold in GAMT deficient mice.
Amino acidsCreatine synthesis and exchanges between brain cells: What can be learned from human creatine deficiencies and various experimental models?
Amino acidsReview: Human guanidinoacetate n-methyl transferase (GAMT) deficiency: A treatable inborn error of metabolism.
Pakistan journal of pharmaceutical sciencesMixed movement disorders revealing an atypical form of creatine deficiency syndrome.
Iranian journal of neurologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de deficiência de creatina.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de deficiência de creatina
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Creatine Deficiency Syndrome: Significance of Magnetic Resonance Spectroscopy.
- Structural insights into the substrate uptake and inhibition of the human creatine transporter (hCRT).Proceedings of the National Academy of Sciences of the United States of America· 2025· PMID 40892912mais citado
- Gene delivery of AGAT and GAMT boosts creatine levels in creatine transporter deficiency patient fibroblasts.
- [Two cases of creatine deficiency syndrome caused by GAMT gene mutations and literature review].Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatrics· 2025· PMID 40105081mais citado
- Imaging the Future: Diagnosing Treatable Neurometabolic Disorders in Children.
- Transporters for Creatine and Related Guanidino Compounds: Their Relevance to Brain Health and Disorders.
- Dynamic electro-clinical features in Guanidinoacetate N-methyltransferase deficiency: A familial case series.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:79172(Orphanet)
- MONDO:0000456(MONDO)
- GARD:18952(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q16908143(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
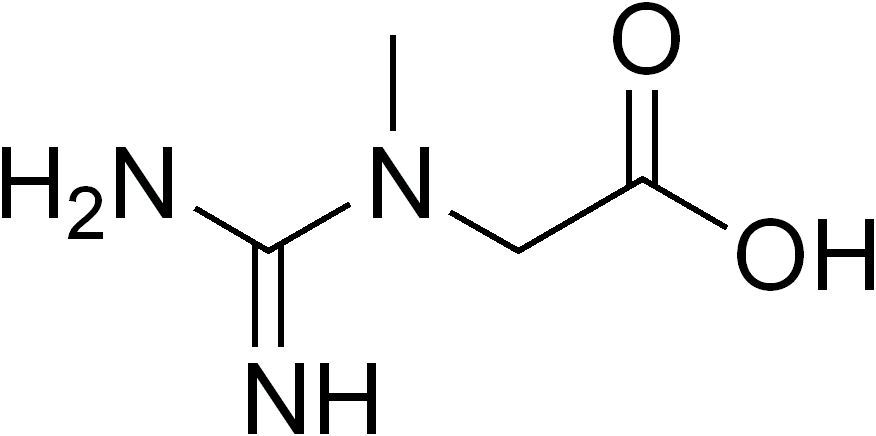
Síndrome de deficiência de creatina
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata