A Deficiência do Transportador de Creatina ligada ao X (CRTR-D) é uma condição causada pela falta de creatina no corpo. Ela se manifesta clinicamente por atraso geral no desenvolvimento e deficiência intelectual (dificuldade de aprendizado), com um atraso notável na fala e na linguagem, comportamentos que lembram o autismo e convulsões.
Introdução
O que você precisa saber de cara
A Deficiência do Transportador de Creatina ligada ao X (CRTR-D) é uma condição causada pela falta de creatina no corpo. Ela se manifesta clinicamente por atraso geral no desenvolvimento e deficiência intelectual (dificuldade de aprendizado), com um atraso notável na fala e na linguagem, comportamentos que lembram o autismo e convulsões.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 26 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 60 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Not applicable, X-linked recessive.
Curadoria gene-doença
fontes oficiaisCreatine:sodium symporter which mediates the uptake of creatine (PubMed:17465020, PubMed:22644605, PubMed:25861866, PubMed:7945388, PubMed:7953292, PubMed:9882430). Plays an important role in supplying creatine to the brain via the blood-brain barrier (By similarity)
Cell membraneApical cell membrane
Cerebral creatine deficiency syndrome 1
An X-linked disorder of creatine transport characterized by intellectual disability, severe speech delay, behavioral abnormalities, and seizures. Carrier females may show mild neuropsychologic impairment.
Variantes genéticas (ClinVar)
566 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Deficiência de transportador da creatina ligada ao X
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
20 ensaios clínicos encontrados, 3 ativos.
Publicações mais relevantes
Longitudinal Characterization of Males With X-Linked Creatine Transporter Deficiency: Final Results of a Multiyear Observational Study.
The purpose of the Vigilan observational study (ClinicalTrials.gov, NCT02931682) was to prospectively assess the natural history and developmental course of creatine transporter deficiency (CTD). Males with CTD aged 6 months to 65 years were evaluated at 6-month intervals for up to 4 years. Evaluations included neurodevelopmental assessments of intellectual functioning, adaptive functioning, challenging behaviors and the onset and progression of medical comorbidities. Fifty participants (median age, 7.6 years) were enrolled. The predominant CTD phenotype consisted of significant intellectual disabilities and limited skill development over time. Most participants had a history of febrile or nonfebrile seizures, gastrointestinal symptoms, and growth failure. All participants learned how to walk, 78% developed at least some verbal speech, and 34% communicated using phrases or sentences. Norm-referenced neurodevelopment assessments indicated declining standardized scores over time; however, absolute scores (i.e., age equivalent person ability scores) indicated that developmental gains were slower than average, particularly among older participants. Between-person differences in neurodevelopmental skills as a function of age did not match within-person change, suggesting a cohort effect. In this cohort, CTD was associated with significant and persistent intellectual disability. The use of absolute metrics from neurodevelopmental tests (e.g., person ability scores) allowed for the quantification of slow, but present, skill development.
Prominent U-waves without QT prolongation in X-linked creatine transporter deficiency caused by SLC6A8 variants.
Creatine transporter deficiency (CTD) is a rare X-linked disease caused by SLC6A8 variants, which impair ATP-dependent energy metabolism in neurons and myocytes. Although the neurologic and muscular manifestations are well characterized, the cardiac phenotype remains poorly understood. Early clinical reports and a transgenic mouse model have raised concerns about possible associations with corrected QT (QTc) interval prolongation and dilated cardiomyopathy. This study aimed to characterize the cardiac phenotype of male patients with CTD. This cross-sectional study prospectively included male patients with CTD with confirmed SLC6A8 pathogenic variants. A systematic cardiological evaluation was performed, including 12-lead resting electrocardiogram (ECG), ambulatory ECG (Holter monitoring), transthoracic echocardiography, and biological analysis. 23 male patients with CTD (median [interquartile range] age 17.1 years [13.5-20.5]) with 20 distinct SLC6A8 variants were included. Prominent U-waves were observed in 82.6% of resting ECGs and 95% of ambulatory ECGs, and biphasic T-waves in 30.4% and 90%, respectively. No patient had a prolonged QTc interval (median [interquartile range] QTc interval 431 ms [411-443]) when the U-waves were excluded. Repolarization abnormalities were not secondary to electrolyte disorders. No sustained arrhythmias or conduction disorders were observed. No patient reported syncope or cardiac arrest. Transthoracic echocardiography revealed no cardiomyopathy or congenital heart defects. 2 patients had mildly elevated N-terminal pro-brain natriuretic peptide with no clinical or imaging abnormalities. This study highlighted an atypical ventricular repolarization pattern in patients with CTD (prominent U-waves and biphasic T-waves) without QTc interval prolongation. Long-term follow-up data are needed to establish its prognosis, but it must be distinguished from long-QT syndrome. No patient met diagnostic criteria for cardiomyopathy or congenital heart defect.
How a patient-led advocacy organization supports the road to diagnosis and treatment of creatine transporter deficiency.
The current era of drug development has evolved significantly. Patient advocacy organizations are moving beyond simply supporting community members and are taking the reins to improve the speed of diagnoses, initiate therapeutic discoveries, and lay the groundwork to ensure successful clinical trials. The Association for Creatine Deficiencies (ACD) is an international parent-led patient advocacy organization focused on the three ultra-rare neurodevelopmental monogenic disorders resulting in Cerebral Creatine Deficiency Syndromes (CCDS). These include X-linked creatine transporter deficiency (CTD), guanidinoacetate methyltransferase (GAMT) deficiency, and l-arginine:glycine amidinotransferase (AGAT) deficiency. While each is rare in its own right, the unified CCDS community is effectively advancing the field of CCDS with each disorder benefiting from progress made in the other two disease areas. ACD collaborators include caregivers, academic researchers, clinicians, industry partners, and policymakers. Since its founding in 2012, the organization has evolved and achieved significant milestones. These include advancements in disease diagnosis, investments in various therapeutic modalities, creation of a collaborative research community, a unified patient community contributing essential patient data, and repositories of patient-derived specimens. The initiatives of ACD are intended to create the earliest diagnosis possible through newborn screening, to have an effective treatment, and to make disease management strategies available to all members of the CCDS community, including those diagnosed at later stages and experiencing greater effects of the diseases.
Arginine, glycine, and creatine supplementation improves symptoms in a female with creatine transporter deficiency.
X-linked creatine transporter deficiency is caused by hemizygous or heterozygous pathogenic variants in SLC6A8 that cause neuropsychiatric symptoms because of impaired uptake of creatine into tissues throughout the body. Small cohorts have suggested that supplementation of creatine, arginine, and glycine can stop disease progression in males, but only six cases of supplementation in females have been published. Here, we present a female with a de-novo pathogenic SLC6A8 variant who had ongoing weight loss, mild intellectual disability, and neuropsychiatric symptoms. Magnetic resonance spectroscopy of the brain showed reduced creatine on all acquired spectra. The patient was started on creatine-monohydrate, l -arginine, and l -glycine supplementation, and she had significant symptomatic improvement within the following 3 weeks. After 8 months of supplementation, magnetic resonance spectroscopy showed improved creatine concentrations with normalizing semiquantitative ratios with other brain metabolites. Current data supports clinicians trialing creatine, arginine, and glycine supplements for female patients with creatine transporter deficiency.
Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review.
(1) Background: X-linked creatine transporter deficiency (CTD) (OMIM 300036) is a rare group of inherited metabolic disorders characterized by global developmental delay/intellectual disability (GDD/ID), seizures, autistic behavior, and movement disorders. Pathogenic variants in the SLC6A8 gene, located at Xq28, are causative of the disease, leading to impaired creatine transport into the brain. Supplementation with creatine and its precursors, glycine and arginine, has been attempted, yet the treatment efficacy remains controversial. (2) Methods: Here we report a de novo SLC6A8 variant in a boy aged 3 years 9 months presenting with GDD, autistic behavior, and epilepsy. Elevated urinary creatine/creatinine ratio and diminished creatine peak on brain MR spectroscopy suggested the diagnosis of CTD. Genetic sequencing revealed a de novo hemizygous frameshift variant (NM_005629: c.1136_1137del, p. Glu379ValfsTer85). Creatine supplementation therapy was initiated after definitive diagnosis. Electroencephalography and MR spectroscopy were monitored during follow-up in concurrence with neuropsychological evaluations. The clinical phenotype and treatment response of CTD were summarized by systematic view of the literature. (3) Results: In silico analysis showed this variant to be deleterious, probably interfering with substrate binding and conformational changes during creatine transport. Creatine supplementation therapy led to seizure cessation and modest cognitive improvement after half-year's treatment. (4) Conclusions: This case highlights the importance of MR spectroscopy and metabolic screening in males with GDD/ID, allowing for early diagnosis and therapeutic intervention. Mechanistic understanding and case-per-se analysis are required to enable precision treatment for the patients.
Publicações recentes
Longitudinal Characterization of Males With X-Linked Creatine Transporter Deficiency: Final Results of a Multiyear Observational Study.
Prominent U-waves without QT prolongation in X-linked creatine transporter deficiency caused by SLC6A8 variants.
How a patient-led advocacy organization supports the road to diagnosis and treatment of creatine transporter deficiency.
Arginine, glycine, and creatine supplementation improves symptoms in a female with creatine transporter deficiency.
Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review.
📚 EuropePMC12 artigos no totalmostrando 15
Longitudinal Characterization of Males With X-Linked Creatine Transporter Deficiency: Final Results of a Multiyear Observational Study.
Pediatric neurologyProminent U-waves without QT prolongation in X-linked creatine transporter deficiency caused by SLC6A8 variants.
Heart rhythmHow a patient-led advocacy organization supports the road to diagnosis and treatment of creatine transporter deficiency.
Frontiers in neuroscienceArginine, glycine, and creatine supplementation improves symptoms in a female with creatine transporter deficiency.
Psychiatric geneticsDiagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review.
Brain sciencesRare disease variant curation from literature: assessing gaps with creatine transport deficiency in focus.
BMC genomicsCase Report: X-Linked Creatine Transporter Deficiency in Two Saudi Brothers with Autism.
Journal of autism and developmental disordersCase report: Clinical and magnetic resonance spectroscopy presentation of a female severely affected with X-linked creatine transporter deficiency.
Radiology case reportsX-linked creatine transporter deficiency results in prolonged QTc and increased sudden death risk in humans and disease model.
Genetics in medicine : official journal of the American College of Medical GeneticsA new rat model of creatine transporter deficiency reveals behavioral disorder and altered brain metabolism.
Scientific reportsCreatine transporter deficiency, an underdiagnosed cause of male intellectual disability.
BMJ case reportsOxidative phosphorylation in creatine transporter deficiency.
NMR in biomedicineA novel SLC6A8 mutation associated with intellectual disabilities in a Chinese family exhibiting creatine transporter deficiency: case report.
BMC medical geneticsVariable White Matter Atrophy and Intellectual Development in a Family With X-linked Creatine Transporter Deficiency Despite Genotypic Homogeneity.
Pediatric neurologyMetabolomic Profiling of Human Urine as a Screen for Multiple Inborn Errors of Metabolism.
Genetic testing and molecular biomarkersAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Deficiência de transportador da creatina ligada ao X.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Deficiência de transportador da creatina ligada ao X
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Longitudinal Characterization of Males With X-Linked Creatine Transporter Deficiency: Final Results of a Multiyear Observational Study.
- Prominent U-waves without QT prolongation in X-linked creatine transporter deficiency caused by SLC6A8 variants.
- How a patient-led advocacy organization supports the road to diagnosis and treatment of creatine transporter deficiency.
- Arginine, glycine, and creatine supplementation improves symptoms in a female with creatine transporter deficiency.
- Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:52503(Orphanet)
- OMIM OMIM:300352(OMIM)
- MONDO:0010305(MONDO)
- GARD:1608(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q17084842(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
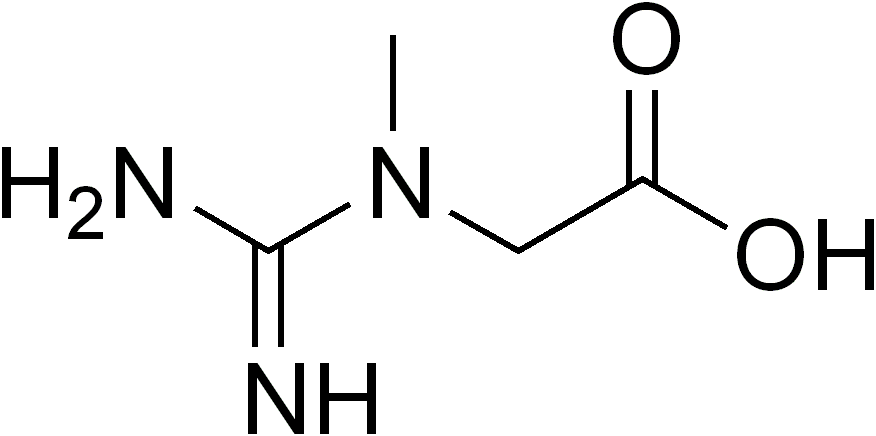
Deficiência de transportador da creatina ligada ao X
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov