Síndrome de duplicação parcial do braço curto do cromossomo 17, caracterizada por prognatismo mandibular, bossas frontais, luxação congênita do quadril e fraqueza diafragmática. Pode apresentar também nariz curto, ventriculomegalia, hérnia inguinal e sintomas neurológicos como parestesia e dor espontânea.
Introdução
O que você precisa saber de cara
Visão geral
A trissomia parcial do braço curto do cromossomo 17 é uma doença genética rara caracterizada pela presença de uma cópia extra de parte do braço curto (braço p) do cromossomo 17. Essa alteração cromossômica pode levar a uma variedade de sinais e sintomas, que variam de pessoa para pessoa. A condição também é conhecida internacionalmente como 'Partial duplication of the short arm of chromosome 17 syndrome'.[1][2]
Sinais e sintomas
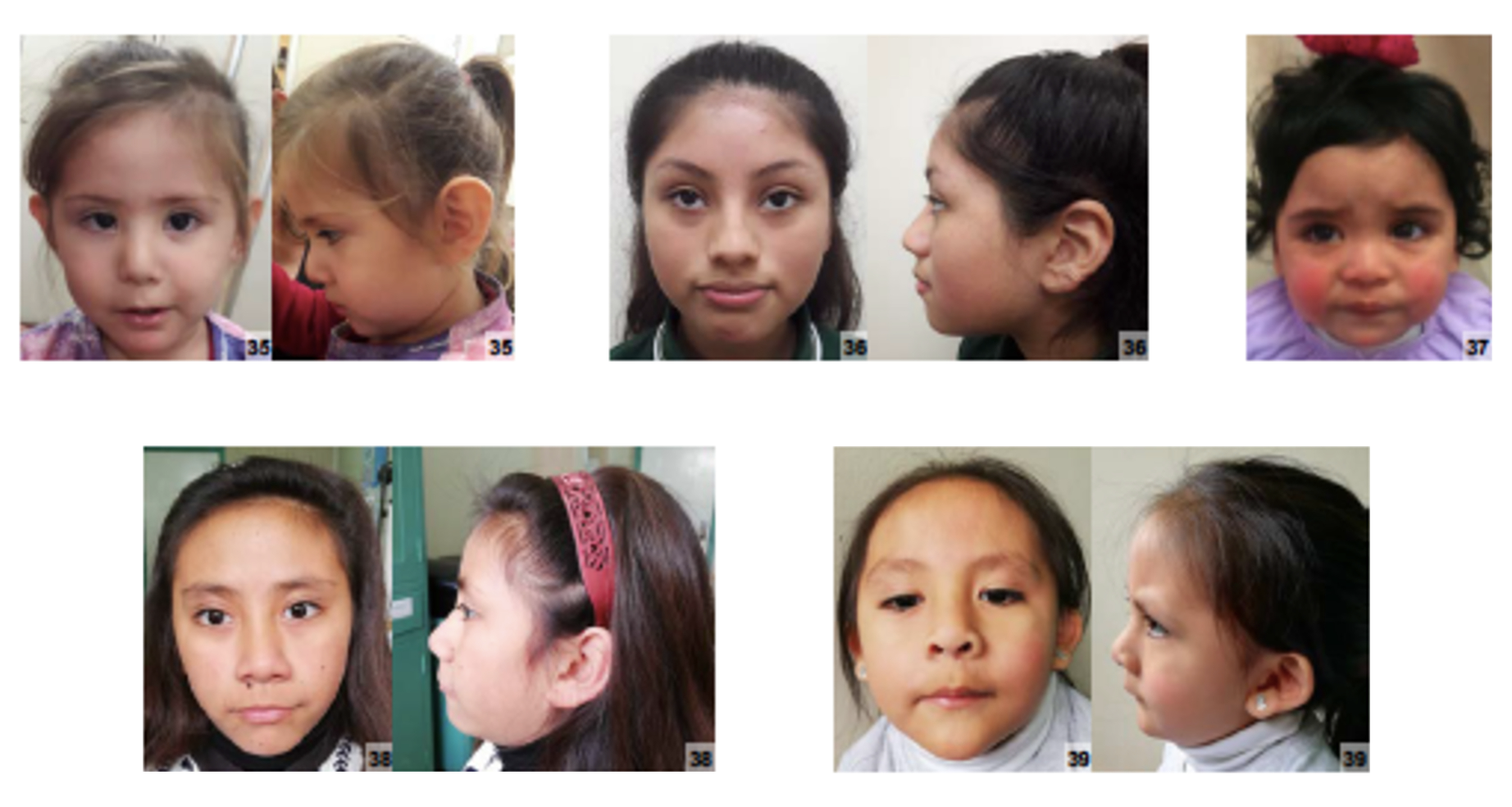
Os sinais e sintomas da trissomia parcial do braço curto do cromossomo 17 podem afetar múltiplos sistemas do corpo. Entre as características físicas, podem ser observados prognatismo mandibular (queixo proeminente), bossas frontais (saliências na testa), testa alta, nariz curto, macroglossia (língua aumentada) e ptose (pálpebra caída). Problemas ósseos e articulares incluem luxação congênita do quadril, cifoescoliose (curvatura anormal da coluna), pé cavo e garra ulnar (deformidade da mão).[1][3]
O sistema nervoso é frequentemente afetado, podendo causar ventriculomegalia (aumento dos ventrículos cerebrais), polineuropatia desmielinizante aguda (danos aos nervos periféricos), neuropatia periférica desmielinizante, parestesia (sensação de formigamento ou dormência), sensação de dor espontânea, fraqueza muscular proximal, fraqueza diafragmática, desequilíbrio da marcha, distúrbio da marcha, ataxia sensorial (falta de coordenação devido à perda de sensibilidade) e reflexos tendinosos profundos hiperativos. Alterações hipertróficas dos nervos e velocidade de condução nervosa motora diminuída também podem estar presentes. Outros achados incluem hérnia inguinal, válvula uretral e parestesia.[1][3]
Causas genéticas
A trissomia parcial do braço curto do cromossomo 17 é causada por uma duplicação de uma parte do braço curto do cromossomo 17. Essa alteração genética pode ocorrer de forma espontânea (nova) ou ser herdada de um dos pais. Quatro genes localizados nessa região são particularmente relevantes para a condição: PMP22 (proteína de membrana peroxissomal 2), PAFAH1B1 (subunidade beta do fator de ativação de plaquetas acetilhidrolase IB), YWHAE (proteína 14-3-3 épsilon) e RAI1 (proteína 1 induzida por ácido retinoico). Acredita-se que a cópia extra desses genes contribua para os sinais e sintomas observados.[1][4]
Diagnóstico
O diagnóstico da trissomia parcial do braço curto do cromossomo 17 é realizado por meio de testes genéticos. Existem 336 testes genéticos disponíveis para detectar essa alteração cromossômica. O ClinVar, um banco de dados público de variantes genéticas, já catalogou 721 variantes associadas a essa condição. O código da doença na ontologia MONDO é MONDO:0016950.[1][4]
Tratamento e manejo
Não há um tratamento específico para a trissomia parcial do braço curto do cromossomo 17. O manejo é focado nos sinais e sintomas apresentados por cada pessoa e pode envolver uma equipe multidisciplinar de especialistas. O objetivo é oferecer suporte e melhorar a qualidade de vida. No Brasil, a condição possui cobertura mínima no SUS, o que significa que alguns procedimentos e consultas podem ser acessados pelo sistema público de saúde, mas a cobertura pode variar.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com trissomia parcial do braço curto do cromossomo 17 é variável e depende da extensão da duplicação cromossômica e dos órgãos e sistemas afetados. O acompanhamento médico regular e o manejo dos sintomas são fundamentais para otimizar a qualidade de vida e o desenvolvimento da pessoa.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome de duplicação parcial do braço curto do cromossomo 17, caracterizada por prognatismo mandibular, bossas frontais, luxação congênita do quadril e fraqueza diafragmática. Pode apresentar também nariz curto, ventriculomegalia, hérnia inguinal e sintomas neurológicos como parestesia e dor espontânea.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A trissomia parcial do braço curto do cromossomo 17 é uma doença genética rara caracterizada pela presença de uma cópia extra de parte do braço curto (braço p) do cromossomo 17. Essa alteração cromossômica pode levar a uma variedade de sinais e sintomas, que variam de pessoa para pessoa. A condição também é conhecida internacionalmente como 'Partial duplication of the short arm of chromosome 17 syndrome'.[1][2]
Sinais e sintomas
Os sinais e sintomas da trissomia parcial do braço curto do cromossomo 17 podem afetar múltiplos sistemas do corpo. Entre as características físicas, podem ser observados prognatismo mandibular (queixo proeminente), bossas frontais (saliências na testa), testa alta, nariz curto, macroglossia (língua aumentada) e ptose (pálpebra caída). Problemas ósseos e articulares incluem luxação congênita do quadril, cifoescoliose (curvatura anormal da coluna), pé cavo e garra ulnar (deformidade da mão).[1][3]
O sistema nervoso é frequentemente afetado, podendo causar ventriculomegalia (aumento dos ventrículos cerebrais), polineuropatia desmielinizante aguda (danos aos nervos periféricos), neuropatia periférica desmielinizante, parestesia (sensação de formigamento ou dormência), sensação de dor espontânea, fraqueza muscular proximal, fraqueza diafragmática, desequilíbrio da marcha, distúrbio da marcha, ataxia sensorial (falta de coordenação devido à perda de sensibilidade) e reflexos tendinosos profundos hiperativos. Alterações hipertróficas dos nervos e velocidade de condução nervosa motora diminuída também podem estar presentes. Outros achados incluem hérnia inguinal, válvula uretral e parestesia.[1][3]
Causas genéticas
A trissomia parcial do braço curto do cromossomo 17 é causada por uma duplicação de uma parte do braço curto do cromossomo 17. Essa alteração genética pode ocorrer de forma espontânea (nova) ou ser herdada de um dos pais. Quatro genes localizados nessa região são particularmente relevantes para a condição: PMP22 (proteína de membrana peroxissomal 2), PAFAH1B1 (subunidade beta do fator de ativação de plaquetas acetilhidrolase IB), YWHAE (proteína 14-3-3 épsilon) e RAI1 (proteína 1 induzida por ácido retinoico). Acredita-se que a cópia extra desses genes contribua para os sinais e sintomas observados.[1][4]
Diagnóstico
O diagnóstico da trissomia parcial do braço curto do cromossomo 17 é realizado por meio de testes genéticos. Existem 336 testes genéticos disponíveis para detectar essa alteração cromossômica. O ClinVar, um banco de dados público de variantes genéticas, já catalogou 721 variantes associadas a essa condição. O código da doença na ontologia MONDO é MONDO:0016950.[1][4]
Tratamento e manejo
Não há um tratamento específico para a trissomia parcial do braço curto do cromossomo 17. O manejo é focado nos sinais e sintomas apresentados por cada pessoa e pode envolver uma equipe multidisciplinar de especialistas. O objetivo é oferecer suporte e melhorar a qualidade de vida. No Brasil, a condição possui cobertura mínima no SUS, o que significa que alguns procedimentos e consultas podem ser acessados pelo sistema público de saúde, mas a cobertura pode variar.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com trissomia parcial do braço curto do cromossomo 17 é variável e depende da extensão da duplicação cromossômica e dos órgãos e sistemas afetados. O acompanhamento médico regular e o manejo dos sintomas são fundamentais para otimizar a qualidade de vida e o desenvolvimento da pessoa.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 65 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 166 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição.
Seems to be involved in pore-forming activity and may contribute to the unspecific permeability of the peroxisomal membrane
Peroxisome membrane
Regulatory subunit (beta subunit) of the cytosolic type I platelet-activating factor (PAF) acetylhydrolase (PAF-AH (I)), an enzyme that catalyzes the hydrolyze of the acetyl group at the sn-2 position of PAF and its analogs and participates in PAF inactivation. Regulates the PAF-AH (I) activity in a catalytic dimer composition-dependent manner (By similarity). Required for proper activation of Rho GTPases and actin polymerization at the leading edge of locomoting cerebellar neurons and postmigra
Cytoplasm, cytoskeletonCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleNucleus membrane
Lissencephaly 1
A classical lissencephaly. It is characterized by agyria or pachygyria and disorganization of the clear neuronal lamination of normal six-layered cortex. The cortex is abnormally thick and poorly organized with 4 primitive layers. Associated with enlarged and dysmorphic ventricles and often hypoplasia of the corpus callosum.
Adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways (PubMed:21189250). Binds to a large number of partners, usually by recognition of a phosphoserine or phosphothreonine motif (PubMed:35343654). Binding generally results in the modulation of the activity of the binding partner (By similarity). Positively regulates phosphorylated protein HSF1 nuclear export to the cytoplasm (PubMed:12917326). Plays a positive role in the antiviral si
NucleusCytoplasmMelanosome
Transcriptional regulator of the circadian clock components: CLOCK, BMAL1, BMAL2, PER1/3, CRY1/2, NR1D1/2 and RORA/C. Positively regulates the transcriptional activity of CLOCK a core component of the circadian clock. Regulates transcription through chromatin remodeling by interacting with other proteins in chromatin as well as proteins in the basic transcriptional machinery. May be important for embryonic and postnatal development. May be involved in neuronal differentiation
CytoplasmNucleus
Smith-Magenis syndrome
Characterized by intellectual disability associated with development and growth delays. Affected persons have characteristic behavioral abnormalities, including self-injurious behaviors and sleep disturbance, and distinct craniofacial and skeletal anomalies.
Variantes genéticas (ClinVar)
721 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
32 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Trissomia parcial do braço curto do cromossomo 17
Centros de Referência SUS
24 centros habilitados pelo SUS para Trissomia parcial do braço curto do cromossomo 17
Centros para Trissomia parcial do braço curto do cromossomo 17
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Potocki-Lupski Syndrome in Ethiopian Child: A Case Report.
Potocki-Lupski syndrome (PTLS) is a rare developmental disorder resulting from the partial duplication of the short arm of chromosome 17. Affected children may have hypotonia, facial dysmorphism, or neurological abnormalities. We present the case of a 5-year-old female patient from Ethiopia diagnosed with Potocki-Lupski syndrome (PTLS)(17p11.2 microduplication) through multiplex ligation-dependent probe amplification (MLPA) testing. This technique identified the duplication of regions of the 17p11.2 chromosome (RAI1, DRC3, USP22, COPS3 and LLGL1). The patient exhibited neurological manifestations including speech delay and mild intellectual disability, along with craniofacial dysmorphism characterized by a triangular face, wide forehead, dental malocclusion, and micrognathia. A multidisciplinary team approach is imperative for managing patients with PTLS. Parental counseling and genetic advice are crucial for families with children affected by PTLS.
Case Report: Potocki-Lupski Syndrome in Five Siblings.
Potocki-Lupski syndrome (PTLS) is a rare developmental disorder resulting from the partial duplication of the short arm of chromosome 17. Affected children may have hypotonia, facial dysmorphism, or neurological abnormalities. PTLS is also frequently associated with failure to thrive due to swallowing difficulties or growth hormone deficiency. We report the first Romanian family (a mother and her five children) diagnosed with PTLS (17p11.2 microduplication). Fortunately, they present a less severe form of the disease. The neurological manifestations (speech delay, mild intellectual disability) are associated with craniofacial dysmorphism (microcephaly, micrognathia, triangular face, broad forehead, long chin, prominent ears, dolichocephaly, down slanting palpebral fissures). The diagnostic was established using a multiplex ligation-dependent probe amplification technique (MLPA) test, which detected the duplication of three regions of the 17p11.2 chromosome (RAI1, DRC3-6, LLGL1-4RA). Children with PTLS have specific phenotypes (craniofacial dysmorphism or neurological manifestations), which must draw the pediatrician's attention to a possible genetic condition. However, every child with this disease is unique and may have a different clinical presentation. A multi-disciplinary team is needed for the management of these patients. The parent's counseling and genetic advice are essential for a family with children with PTLS.
Partial trisomy 4q and monosomy 5p inherited from a maternal translocationt(4;5)(q33; p15) in three adverse pregnancies.
Carriers of balanced reciprocal chromosomal translocations are at known reproductive risk for offspring with unbalanced genotypes and resultantly abnormal phenotypes. Once fertilization of a balanced translocation gamete with a normal gamete, the partial monosomy or partial trisomy embryo will undergo abortion, fetal arrest or fetal malformations. We reported a woman with chromosomal balanced translocation who had two adverse pregnancies. Prenatal diagnosis was made for her third pregnancy to provide genetic counseling and guide her fertility. We presented a woman with chromosomal balanced translocation who had three adverse pregnancies. Routine G banding and CNV-seq were used to analyze the chromosome karyotypes and copy number variants of amniotic fluid cells and peripheral blood. The karyotype of the woman was 46,XX,t(4;5)(q33;p15). During her first pregnancy, odinopoeia was performed due to fetal edema and abdominal fluid. The umbilical cord tissue of the fetus was examined by CNV-seq. The results showed a genomic gain of 24.18 Mb at 4q32.3-q35.2 and a genomic deletion of 10.84 Mb at 5p15.2-p15.33 and 2.36 Mb at 15q11.1-q11.2. During her second pregnancy, she did not receive a prenatal diagnosis because a routine prenatal ultrasound examination found no abnormalities. In 2016, she gave birth to a boy. The karyotype the of the boy was 46,XY,der(5)t(4;5)(q33;p15)mat. The results of CNV-seq showed a deletion of short arm of chromosome 5 capturing regions 5p15.2-p15.33, a copy gain of the distal region of chromosome 4 at segment 4q32.3q35.2, a duplication of chromosome 1 at segment 1q41q42.11 and a duplication of chromosome 17 at segment 17p12. During her third pregnancy, she underwent amniocentesis at 17 weeks of gestation. Chromosome karyotype hinted 46,XY,der(5)t(4;5)(q33;p15)mat. Results of CNV-seq showed a deletion of short arm (p) of chromosome 5 at the segment 5p15.2p15.33 and a duplication of the distal region of chromosome 4 at segment 4q32.3q35.2. Chromosomal abnormalities in three pregnancies were inherited from the mother. Preimplantation genetic diagnosis is recommended to prevent the birth of children with chromosomal abnormalities.
Neuronal migration genes and a familial translocation t (3;17): candidate genes implicated in the phenotype.
While Miller-Dieker syndrome critical region deletions are well known delineated anomalies, submicroscopic duplications in this region have recently emerged as a new distinctive syndrome. So far, only few cases have been described overlapping 17p13.3 duplications. In this study, we report on clinical and cytogenetic characterization of two new cases involving 17p13.3 and 3p26 chromosomal regions in two sisters with familial history of lissencephaly. Fluorescent In Situ Hybridization and array Comparative Genomic Hybridization were performed. A deletion including the critical region of the Miller-Dieker syndrome of at least 2,9 Mb and a duplication of at least 3,6 Mb on the short arm of chromosome 3 were highlighted in one case. The opposite rearrangements, 17p13.3 duplication and 3p deletion, were observed in the second case. This double chromosomal aberration is the result of an adjacent 1:1 meiotic segregation of a maternal reciprocal translocation t(3,17)(p26.2;p13.3). 17p13.3 and 3p26 deletions have a clear range of phenotypic features while duplications still have an uncertain clinical significance. However, we could suggest that regardless of the type of the rearrangement, the gene dosage and interactions of CNTN4, CNTN6 and CHL1 in the 3p26 and PAFAH1B1, YWHAE in 17p13.3 could result in different clinical spectrums.
Publicações recentes
Neuronal migration genes and a familial translocation t (3;17): candidate genes implicated in the phenotype.
A child with split-hand/foot associated with tibial hemimelia (SHFLD syndrome) and thrombocytopenia maps to chromosome region 17p13.3.
Neuroblastoma in a dysmorphic girl with a partial duplication of 2p caused by an unbalanced translocation.
DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome.
A molecular, cytogenetic, and clinical evaluation of mosaic tandem duplication 17p and Charcot-Marie-Tooth type 1A neuropathy.
📚 EuropePMCmostrando 4
Potocki-Lupski Syndrome in Ethiopian Child: A Case Report.
Pediatric health, medicine and therapeuticsCase Report: Potocki-Lupski Syndrome in Five Siblings.
Frontiers in pediatricsPartial trisomy 4q and monosomy 5p inherited from a maternal translocationt(4;5)(q33; p15) in three adverse pregnancies.
Molecular cytogeneticsNeuronal migration genes and a familial translocation t (3;17): candidate genes implicated in the phenotype.
BMC medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Trissomia parcial do braço curto do cromossomo 17.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Trissomia parcial do braço curto do cromossomo 17
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Potocki-Lupski Syndrome in Ethiopian Child: A Case Report.
- Case Report: Potocki-Lupski Syndrome in Five Siblings.
- Partial trisomy 4q and monosomy 5p inherited from a maternal translocationt(4;5)(q33; p15) in three adverse pregnancies.
- Neuronal migration genes and a familial translocation t (3;17): candidate genes implicated in the phenotype.
- A child with split-hand/foot associated with tibial hemimelia (SHFLD syndrome) and thrombocytopenia maps to chromosome region 17p13.3.
- Neuroblastoma in a dysmorphic girl with a partial duplication of 2p caused by an unbalanced translocation.
- DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome.
- A molecular, cytogenetic, and clinical evaluation of mosaic tandem duplication 17p and Charcot-Marie-Tooth type 1A neuropathy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:262803(Orphanet)
- MONDO:0016950(MONDO)
- GARD:20870(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55786659(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Trissomia parcial do braço curto do cromossomo 17
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata