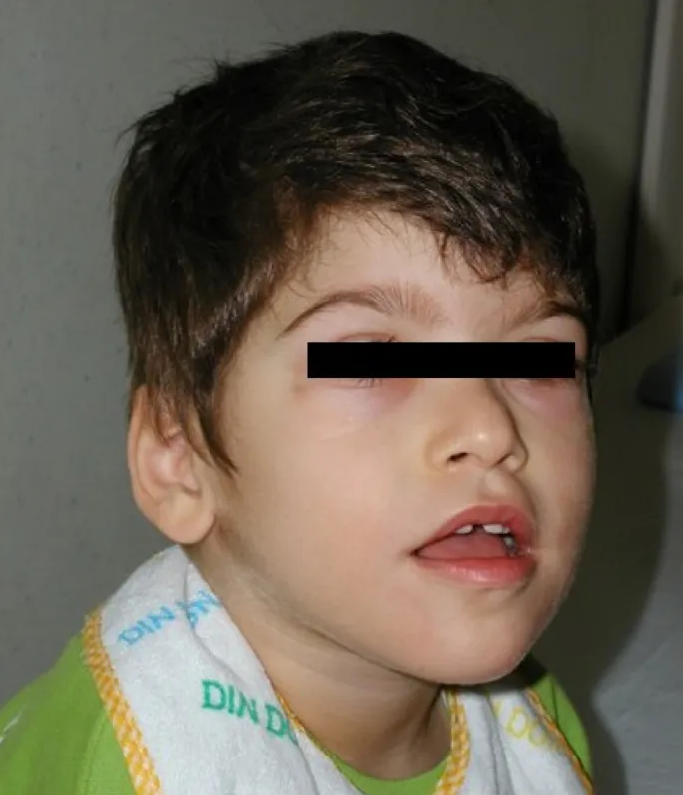
A síndrome de Feingold tipo 1 (FS1) é uma síndrome de malformação hereditária rara caracterizada por microcefalia, baixa estatura e numerosas anomalias digitais.
Introdução
O que você precisa saber de cara
A síndrome de Feingold tipo 1 (FS1) é uma síndrome de malformação hereditária rara caracterizada por microcefalia, baixa estatura e numerosas anomalias digitais.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 20 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 65 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisPositively regulates the transcription of MYCNOS in neuroblastoma cells
Nucleus
Variantes genéticas (ClinVar)
145 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 123 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de Feingold, tipo 1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Rare features in Feingold syndrome type 1.
Feingold syndrome type 1 (FS1) (OMIM 164280) is an autosomal dominant condition due to heterozygous loss of function variants in MYCN gene or to 2p24 deletion encompassing MYCN gene. The core features of FS1 are digital anomalies, microcephaly, facial dysmorphism, short stature, esophageal/duodenal atresia, and mild learning disabilities. Additional features are reported in a minority of patients, such as cardiac and renal anomalies. Sensorineural deafness is reported in 7 % of the patients. Other features can be associated with classical features of FS1 in patients with 2p deletion including MYCN and other genes. Recently, absence of the flexor pollicis longus tendon has been reported as a new skeletal feature in a pedigree segregating a MYCN variant. Here, we reported on three patients having FS1 without gastrointestinal atresia and unusual features: laryngeal cleft, congenital deafness, agenesis of the corpus callosum, and radio ulnar-synostosis (RUS). After the extension of the genetic screening, RUS was considered as an independent condition linked to SMAD6 variant. Diagnosis of FS1 can be challenging when there are unusual features without digestive malformations drawing attention. In this situation, the diagnostic approach may be based on major criteria of FS1: i) brachymesophalangy of the 2nd and 5th fingers, brachydactyly of fingers and toes with or without 2/3 and/or 4/5 toe syndactylies, ii) microcephaly, and iii) radiographs of the feet to look for amesophalangy of toes. Extension of the genetic screening is required to eliminate the possibility of two independent conditions. In addition to the previous recommendations, we advocate for a set of recommendations for evaluation of FS1 patients following initial diagnosis: systematic search of deafness, verification of the flexion of the interphalangeal joints of the thumbs, laryngoscopy in case of stridor or swallowing disorders, and finally systematic cerebral MRI.
Feingold syndrome with GJB2 variants.
Congenital hearing loss is the most common birth defect, with genetic factors implicated in 50 % of prelingual cases. GJB2 variant, causing up to 50 % of autosomal recessive non-syndromic hearing loss, typically show stable hearing profiles and favorable cochlear implant (CI) outcomes. In this case, however, the atypical clinical course prompted further evaluation. A 3-year-6-month-old girl, born at 35 weeks presented with profound bilateral hearing loss detected by auditory brainstem response. The patient had normal tympanic membranes, angulated ears, microcephaly, short stature, narrow palpebral fissures, and digital anomalies. CT revealed inner ear malformations including bilateral vestibular enlargement and cochlear nerve canal stenosis. Genetic testing showed a homozygous GJB2 c.235delC (p.L79fs) variant. Initial hearing aids proved insufficient, leading to CI placement, which improved hearing thresholds. Language and social delays persisted. As her older sister, without a GJB2 variant, had hearing loss and a family history of characteristic physical symptoms, further genetic analysis revealed a heterozygous MYCN variant (NM_005378:c.1138_1139del:p.S380fs), confirming Feingold syndrome type 1 (FS1). This report is a very rare report of FS 1 combined with severe hearing loss due to a GJB2 variant. Early screening for malformations not detected by GJB2 led to accurate diagnosis and provision of information to the family.
MYCN in human development and diseases.
Somatic mutations in MYCN have been identified across various tumors, playing pivotal roles in tumorigenesis, tumor progression, and unfavorable prognoses. Despite its established notoriety as an oncogenic driver, there is a growing interest in exploring the involvement of MYCN in human development. While MYCN variants have traditionally been associated with Feingold syndrome type 1, recent discoveries highlight gain-of-function variants, specifically p.(Thr58Met) and p.(Pro60Leu), as the cause for megalencephaly-polydactyly syndrome. The elucidation of cellular and murine analytical data from both loss-of-function (Feingold syndrome model) and gain-of-function models (megalencephaly-polydactyly syndrome model) is significantly contributing to a comprehensive understanding of the physiological role of MYCN in human development and pathogenesis. This review discusses the MYCN's functional implications for human development by reviewing the clinical characteristics of these distinct syndromes, Feingold syndrome, and megalencephaly-polydactyly syndrome, providing valuable insights into the understanding of pathophysiological backgrounds of other syndromes associated with the MYCN pathway and the overall comprehension of MYCN's role in human development.
Feingold syndrome type 1: a rare cause of fetal microcephaly (prenatal diagnosis).
We report a case of fetal microcephaly found during the second trimester ultrasound and confirmed by further ultrasound scans and fetal MRI. The array comparative genomic hybridisation analysis of the fetus and the male parent showed a 1.5 Mb deletion overlapping the Feingold syndrome region, an autosomal dominant syndrome that can cause microcephaly, facial/hand abnormalities, mild neurodevelopmental delay and others. This case illustrates the need for a detailed investigation by a multidisciplinary team to provide prenatal counselling regarding a postnatal outcome to the parents and orient their decision towards the continuation or termination of pregnancy.
Mycn regulates intestinal development through ribosomal biogenesis in a zebrafish model of Feingold syndrome 1.
Feingold syndrome type 1, caused by loss-of-function of MYCN, is characterized by varied phenotypes including esophageal and duodenal atresia. However, no adequate model exists for studying the syndrome's pathological or molecular mechanisms, nor is there a treatment strategy. Here, we developed a zebrafish Feingold syndrome type 1 model with nonfunctional mycn, which had severe intestinal atresia. Single-cell RNA-seq identified a subcluster of intestinal cells that were highly sensitive to Mycn, and impaired cell proliferation decreased the overall number of intestinal cells in the mycn mutant fish. Bulk RNA-seq and metabolomic analysis showed that expression of ribosomal genes was down-regulated and that amino acid metabolism was abnormal. Northern blot and ribosomal profiling analysis showed abnormal rRNA processing and decreases in free 40S, 60S, and 80S ribosome particles, which led to impaired translation in the mutant. Besides, both Ribo-seq and western blot analysis showed that mTOR pathway was impaired in mycn mutant, and blocking mTOR pathway by rapamycin treatment can mimic the intestinal defect, and both L-leucine and Rheb, which can elevate translation via activating TOR pathway, could rescue the intestinal phenotype of mycn mutant. In summary, by this zebrafish Feingold syndrome type 1 model, we found that disturbance of ribosomal biogenesis and blockage of protein synthesis during development are primary causes of the intestinal defect in Feingold syndrome type 1. Importantly, our work suggests that leucine supplementation may be a feasible and easy treatment option for this disease.
Publicações recentes
Rare features in Feingold syndrome type 1.
🥉 Relato de casoFeingold syndrome with GJB2 variants.
MYCN in human development and diseases.
Feingold syndrome type 1: a rare cause of fetal microcephaly (prenatal diagnosis).
Mycn regulates intestinal development through ribosomal biogenesis in a zebrafish model of Feingold syndrome 1.
📚 EuropePMC48 artigos no totalmostrando 7
Rare features in Feingold syndrome type 1.
European journal of medical geneticsFeingold syndrome with GJB2 variants.
Auris, nasus, larynxMYCN in human development and diseases.
Frontiers in oncologyFeingold syndrome type 1: a rare cause of fetal microcephaly (prenatal diagnosis).
BMJ case reportsMycn regulates intestinal development through ribosomal biogenesis in a zebrafish model of Feingold syndrome 1.
PLoS biologyClinical and molecular characterizations of 11 new patients with type 1 Feingold syndrome: Proposal for selecting diagnostic criteria and further genetic testing in patients with severe phenotype.
American journal of medical genetics. Part ADistinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models.
Nature communicationsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de Feingold, tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de Feingold, tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Rare features in Feingold syndrome type 1.
- Feingold syndrome with GJB2 variants.
- MYCN in human development and diseases.
- Feingold syndrome type 1: a rare cause of fetal microcephaly (prenatal diagnosis).
- Mycn regulates intestinal development through ribosomal biogenesis in a zebrafish model of Feingold syndrome 1.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:391641(Orphanet)
- OMIM OMIM:164280(OMIM)
- MONDO:0008115(MONDO)
- GARD:17624(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de Feingold, tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)