A Síndrome Orelha-Patela-Baixa Estatura é um conjunto de problemas que inclui microtia bilateral (orelhas muito pequenas ou malformadas), ausência das rótulas (os ossos dos joelhos), baixa estatura, dificuldade para ganhar peso e traços faciais marcantes, como testa alta, queixo pequeno, lábios cheios, boca pequena e sulcos nasolabiais acentuados (as "rugas do sorriso" que ligam as narinas aos cantos da boca).
Introdução
O que você precisa saber de cara
A Síndrome Orelha-Patela-Baixa Estatura é um conjunto de problemas que inclui microtia bilateral (orelhas muito pequenas ou malformadas), ausência das rótulas (os ossos dos joelhos), baixa estatura, dificuldade para ganhar peso e traços faciais marcantes, como testa alta, queixo pequeno, lábios cheios, boca pequena e sulcos nasolabiais acentuados (as "rugas do sorriso" que ligam as narinas aos cantos da boca).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 68 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 180 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
8 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Curadoria gene-doença
fontes oficiaisMitochondrial ornithine-citrulline antiporter (Probable) (PubMed:12807890, PubMed:22262851). Catalyzes the exchange between cytosolic ornithine and mitochondrial citrulline plus an H(+), the proton compensates the positive charge of ornithine thus leading to an electroneutral transport. Plays a crucial role in the urea cycle, by connecting the cytosolic and the intramitochondrial reactions of the urea cycle (Probable) (PubMed:12807890, PubMed:22262851). Lysine and arginine are also transported b
Mitochondrion inner membraneMitochondrion membrane
Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome
An autosomal recessive disorder of the urea cycle characterized by onset in early life. The acute phase of the disease is characterized by vomiting, ataxia, lethargy, confusion, and coma. Chronic clinical manifestations include hypotonia, developmental delay, progressive encephalopathy with mental regression, and spastic paraparesis with pyramidal signs.
Acts as a component of the MCM2-7 complex (MCM complex) which is the replicative helicase essential for 'once per cell cycle' DNA replication initiation and elongation in eukaryotic cells (PubMed:40940420). Core component of CDC45-MCM-GINS (CMG) helicase, the molecular machine that unwinds template DNA during replication, and around which the replisome is built (PubMed:16899510, PubMed:32453425, PubMed:34694004, PubMed:34700328, PubMed:35585232). The active ATPase sites in the MCM2-7 ring are fo
NucleusChromosome
Meier-Gorlin syndrome 8
A form of Meier-Gorlin syndrome, a syndrome characterized by bilateral microtia, aplasia/hypoplasia of the patellae, and severe intrauterine and postnatal growth retardation with short stature and poor weight gain. Additional clinical findings include anomalies of cranial sutures, microcephaly, apparently low-set and simple ears, microstomia, full lips, highly arched or cleft palate, micrognathia, genitourinary tract anomalies, and various skeletal anomalies. While almost all cases have primordial dwarfism with substantial prenatal and postnatal growth retardation, not all cases have microcephaly, and microtia and absent/hypoplastic patella are absent in some. Despite the presence of microcephaly, intellect is usually normal. MGORS8 inheritance is autosomal recessive.
Required for initiation of chromosomal DNA replication. Core component of CDC45-MCM-GINS (CMG) helicase, the molecular machine that unwinds template DNA during replication, and around which the replisome is built
NucleusChromosome
Meier-Gorlin syndrome 7
A form of Meier-Gorlin syndrome, a syndrome characterized by bilateral microtia, aplasia/hypoplasia of the patellae, and severe intrauterine and postnatal growth retardation with short stature and poor weight gain. Additional clinical findings include anomalies of cranial sutures, microcephaly, apparently low-set and simple ears, microstomia, full lips, highly arched or cleft palate, micrognathia, genitourinary tract anomalies, and various skeletal anomalies. While almost all cases have primordial dwarfism with substantial prenatal and postnatal growth retardation, not all cases have microcephaly, and microtia and absent/hypoplastic patella are absent in some. Despite the presence of microcephaly, intellect is usually normal. MGORS7 inheritance is autosomal recessive.
Involved in the initiation of DNA replication. Also participates in checkpoint controls that ensure DNA replication is completed before mitosis is initiated
NucleusCytoplasm
Meier-Gorlin syndrome 5
A syndrome characterized by bilateral microtia, aplasia/hypoplasia of the patellae, and severe intrauterine and postnatal growth retardation with short stature and poor weight gain. Additional clinical findings include anomalies of cranial sutures, microcephaly, apparently low-set and simple ears, microstomia, full lips, highly arched or cleft palate, micrognathia, genitourinary tract anomalies, and various skeletal anomalies. While almost all cases have primordial dwarfism with substantial prenatal and postnatal growth retardation, not all cases have microcephaly, and microtia and absent/hypoplastic patella are absent in some. Despite the presence of microcephaly, intellect is usually normal.
Component of the origin recognition complex (ORC) that binds origins of replication. DNA-binding is ATP-dependent. The specific DNA sequences that define origins of replication have not been identified yet. ORC is required to assemble the pre-replication complex necessary to initiate DNA replication. Binds histone H3 and H4 trimethylation marks H3K9me3, H3K27me3 and H4K20me3
Nucleus
Meier-Gorlin syndrome 2
A syndrome characterized by bilateral microtia, aplasia/hypoplasia of the patellae, and severe intrauterine and postnatal growth retardation with short stature and poor weight gain. Additional clinical findings include anomalies of cranial sutures, microcephaly, apparently low-set and simple ears, microstomia, full lips, highly arched or cleft palate, micrognathia, genitourinary tract anomalies, and various skeletal anomalies. While almost all cases have primordial dwarfism with substantial prenatal and postnatal growth retardation, not all cases have microcephaly, and microtia and absent/hypoplastic patella are absent in some. Despite the presence of microcephaly, intellect is usually normal.
Component of the origin recognition complex (ORC) that binds origins of replication. DNA-binding is ATP-dependent. The specific DNA sequences that define origins of replication have not been identified yet. ORC is required to assemble the pre-replication complex necessary to initiate DNA replication. Does not bind histone H3 and H4 trimethylation marks H3K9me3, H3K27me3 and H4K20me3
Nucleus
Meier-Gorlin syndrome 3
A syndrome characterized by bilateral microtia, aplasia/hypoplasia of the patellae, and severe intrauterine and postnatal growth retardation with short stature and poor weight gain. Additional clinical findings include anomalies of cranial sutures, microcephaly, apparently low-set and simple ears, microstomia, full lips, highly arched or cleft palate, micrognathia, genitourinary tract anomalies, and various skeletal anomalies. While almost all cases have primordial dwarfism with substantial prenatal and postnatal growth retardation, not all cases have microcephaly, and microtia and absent/hypoplastic patella are absent in some. Despite the presence of microcephaly, intellect is usually normal.
Inhibits DNA replication by preventing the incorporation of MCM complex into pre-replication complex (pre-RC) (PubMed:14993212, PubMed:20129055, PubMed:24064211, PubMed:9635433). It is degraded during the mitotic phase of the cell cycle (PubMed:14993212, PubMed:24064211, PubMed:9635433). Its destruction at the metaphase-anaphase transition permits replication in the succeeding cell cycle (PubMed:14993212, PubMed:24064211, PubMed:9635433). Inhibits histone acetyltransferase activity of KAT7/HBO1
CytoplasmNucleus
Meier-Gorlin syndrome 6
A form of Meier-Gorlin syndrome, a syndrome characterized by bilateral microtia, aplasia/hypoplasia of the patellae, and severe intrauterine and postnatal growth retardation with short stature and poor weight gain. Additional clinical findings include anomalies of cranial sutures, microcephaly, apparently low-set and simple ears, microstomia, full lips, highly arched or cleft palate, micrognathia, genitourinary tract anomalies, and various skeletal anomalies. While almost all cases have primordial dwarfism with substantial prenatal and postnatal growth retardation, not all cases have microcephaly, and microtia and absent/hypoplastic patella are absent in some. Despite the presence of microcephaly, intellect is usually normal.
Required for both DNA replication and mitosis (PubMed:11125146, PubMed:14993212, PubMed:21856198, PubMed:22581055, PubMed:26842564). DNA replication licensing factor, required for pre-replication complex assembly. Cooperates with CDC6 and the origin recognition complex (ORC) during G1 phase of the cell cycle to promote the loading of the mini-chromosome maintenance (MCM) complex onto DNA to generate pre-replication complexes (pre-RC) (PubMed:14672932). Required also for mitosis by promoting stab
NucleusChromosome, centromere, kinetochore
Meier-Gorlin syndrome 4
A syndrome characterized by bilateral microtia, aplasia/hypoplasia of the patellae, and severe intrauterine and postnatal growth retardation with short stature and poor weight gain. Additional clinical findings include anomalies of cranial sutures, microcephaly, apparently low-set and simple ears, microstomia, full lips, highly arched or cleft palate, micrognathia, genitourinary tract anomalies, and various skeletal anomalies. While almost all cases have primordial dwarfism with substantial prenatal and postnatal growth retardation, not all cases have microcephaly, and microtia and absent/hypoplastic patella are absent in some. Despite the presence of microcephaly, intellect is usually normal.
Variantes genéticas (ClinVar)
499 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
15 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome orelha-rótula-baixa estatura
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome.
High-throughput sequencing has become a standard first-tier approach for both diagnostics and research-based genetic testing. Consequently, this hypothesis-free testing manner has revealed the true breadth of clinical features for many established genetic disorders, including Meier-Gorlin syndrome (MGORS). Previously known as ear-patella short stature syndrome, MGORS is characterized by growth delay, microtia, and patella hypo/aplasia, as well as genital abnormalities, and breast agenesis in females. Following the initial identification of genetic causes in 2011, a total of 13 genes have been identified to date associated with MGORS. In this review, we summarise the genetic and clinical findings of each gene associated with MGORS and highlight molecular insights that have been made through studying patient variants. We note interesting observations arising across this group of genes as the number of patients has increased, such as the unusually high number of synonymous variants affecting splicing in CDC45 and a subgroup of genes that also cause craniosynostosis. We focus on the complicated molecular genetics for DONSON, where we examine potential genotype-phenotype patterns using the first 3D structural model of DONSON. The canonical role of all proteins associated with MGORS are involved in different stages of DNA replication and in addition to summarising how patient variants impact on this process, we discuss the potential contribution of non-canonical roles of these proteins to the pathophysiology of MGORS.
Novel Compound Heterozygous Variants in the CDC6 Gene in a Russian Patient with Meier-Gorlin Syndrome.
Meier-Gorlin syndrome (MGS) is a rare genetic syndrome inherited in an autosomal dominant or autosomal recessive manner. The disorder is characterized by bilateral microtia, absence or hypoplasia of the patella, and an intrauterine growth retardation as well as a number of other characteristic features. The cause of the disease is mutations in genes encoding proteins involved in the regulation of the cell cycle (ORC1, ORC4, ORC6, CDT1, CDC6, GMNN, CDC45L, MCM3, MCM5, MCM7, GINS2, and DONSON). Meier-Gorlin syndrome 5 due to mutations in the CDC6 gene is difficult to diagnose, and few clinical data have been described to date. Only one patient (male) with a missense mutation in a homozygous state has been previously reported. This report describes a new clinical case of Meier-Gorlin syndrome 5. This is also the first report of a Russian patient with Meier-Gorlin syndrome. The patient, a female, had extremely low physical development, neonatal progeroid appearance, lipodystrophy, thin skin, partial alopecia, cyanosis of the face, triangular face, microgenia, arachnodactyly, delayed bone age, hepatomegaly, hypoplasia of the labia majora, and hypertrophy of the clitoris in addition to known clinical signs. Differential diagnosis was performed with chromosomal abnormalities and Hutchinson-Gilford progeria. According to the results of sequencing of the clinical exome, the patient had two previously undescribed variants in the CDC6 gene, c.230A>G (p.(Lys77Arg)) and c.232C>T (p.(Gln78Ter)), NM_001254.3, in a compound heterozygous state. This case allows us to learn more about the clinical features and nature of MGS 5 and improve the speed of diagnostics and quality of genetic counseling for such families.
Publicações recentes
The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome.
Novel Compound Heterozygous Variants in the CDC6 Gene in a Russian Patient with Meier-Gorlin Syndrome.
Left cerebral hemisphere and ventricular system abnormalities in a Mexican Meier Gorlin syndrome patient: widening the clinical spectrum.
Meier-Gorlin (ear-patella-short stature) syndrome: growth hormone deficiency and previously unrecognized findings.
Meier-Gorlin syndrome (ear-patella-short stature syndrome) in an Italian patient: clinical evaluation and analysis of possible candidate genes.
📚 EuropePMC7 artigos no totalmostrando 2
The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome.
European journal of human genetics : EJHGNovel Compound Heterozygous Variants in the CDC6 Gene in a Russian Patient with Meier-Gorlin Syndrome.
The application of clinical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome orelha-rótula-baixa estatura.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome orelha-rótula-baixa estatura
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome.
- Novel Compound Heterozygous Variants in the CDC6 Gene in a Russian Patient with Meier-Gorlin Syndrome.
- Left cerebral hemisphere and ventricular system abnormalities in a Mexican Meier Gorlin syndrome patient: widening the clinical spectrum.
- Meier-Gorlin (ear-patella-short stature) syndrome: growth hormone deficiency and previously unrecognized findings.
- Meier-Gorlin syndrome (ear-patella-short stature syndrome) in an Italian patient: clinical evaluation and analysis of possible candidate genes.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2554(Orphanet)
- MONDO:0016817(MONDO)
- GARD:2033(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q19587381(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
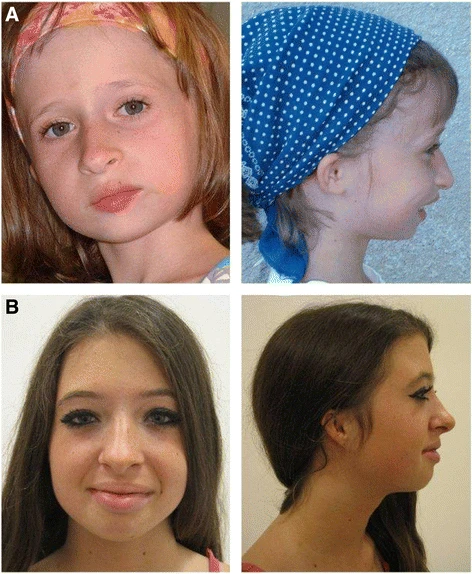
Síndrome orelha-rótula-baixa estatura
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata