A síndrome da sinostose rádio-ulnar-trombocitopenia amegacariocítica é caracterizada pela associação de fusão proximal do rádio e da ulna com trombocitopenia amegacariocítica congênita. Menos de 10 casos foram relatados na literatura até o momento. A síndrome é transmitida como traço autossômico dominante e é causada por mutações no gene HOXA11 (7p15).
Introdução
O que você precisa saber de cara
A síndrome da sinostose rádio-ulnar-trombocitopenia amegacariocítica é caracterizada pela associação de fusão proximal do rádio e da ulna com trombocitopenia amegacariocítica congênita. Menos de 10 casos foram relatados na literatura até o momento. A síndrome é transmitida como traço autossômico dominante e é causada por mutações no gene HOXA11 (7p15).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 8 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 28 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Functions as a transcriptional regulator binding to DNA sequences in the promoter region of target genes and regulating positively or negatively their expression. Oncogene which plays a role in development, cell proliferation and differentiation. May also play a role in apoptosis through regulation of the JNK and TGF-beta signaling. Involved in hematopoiesis Displays histone methyltransferase activity and monomethylates 'Lys-9' of histone H3 (H3K9me1). Probably catalyzes the monomethylation of f
NucleusNucleus speckleCytoplasm
Sequence-specific transcription factor which is part of a developmental regulatory system that provides cells with specific positional identities on the anterior-posterior axis
Nucleus
Radioulnar synostosis with amegakaryocytic thrombocytopenia 1
The syndrome consists of an unusual association of bone marrow failure and skeletal defects. Patients have the same skeletal defects, the proximal fusion of the radius and ulna, resulting in extremely limited pronation and supination of the forearm. Some patients have also symptomatic thrombocytopenia, with bruising and bleeding problems since birth, necessitating correction by bone marrow or umbilical-cord stem-cell transplantation.
Variantes genéticas (ClinVar)
141 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de sinostose radio-cubital-trombocitopenia amegacariocítica
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Reduced-intensity conditioning is effective for allogeneic hematopoietic stem cell transplantation in infants with MECOM-associated syndrome.
Mutations in the MECOM encoding EVI1 are observed in infants who have radioulnar synostosis with amegakaryocytic thrombocytopenia. MECOM-associated syndrome was proposed based on clinical heterogeneity. Allogeneic hematopoietic stem cell transplantation (HSCT) is a curative treatment for progressive bone marrow failure. However, data regarding allogeneic HSCT for this rare disease are limited. We retrospectively assessed overall survival, conditioning regimen, regimen-related toxicities and long-term sequelae in six patients treated with allogeneic HSCT. All patients received a reduced-intensity conditioning (RIC) regimen consisting of fludarabine, cyclophosphamide or melphalan, and rabbit anti-thymocyte globulin and/or low-dose total body/thoracic-abdominal/total lymphoid irradiation, followed by allogeneic bone marrow or cord blood transplantation from unrelated donors between 4 and 18 months of age. All patients survived and achieved stable engraftment and complete chimerization with the donor type. Moreover, no patient experienced severe regimen-related toxicities, and only lower grades of acute graft-versus-host disease were observed. Three patients treated with low-dose irradiation had relatively short stature compared to three patients not treated with irradiation. Therefore, allogeneic HSCT with RIC is an effective and feasible treatment for infants with MECOM-associated syndrome. Future studies are needed to evaluate the use of low-dose irradiation to avoid risks of other long-term sequelae.
Congenital amegakaryocytic thrombocytopenia - Not a single disease.
Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare inherited bone marrow failure syndrome (IBMFS) that is characterized by severe thrombocytopenia at birth due to ineffective megakaryopoiesis and development towards aplastic anemia during the first years of life. CAMT is not a single monogenetic disorder; rather, many descriptions of CAMT include different entities with different etiologies. CAMT in a narrow sense, which is primarily restricted to the hematopoietic system, is caused mainly by mutations in the gene for the thrombopoietin receptor (MPL), sometimes in the gene for its ligand (THPO). CAMT in association with radio-ulnar synostosis, which is not always clinically apparent, is mostly caused by mutations in MECOM, rarely in HOXA11. Patients affected by other IBMFS - especially Fanconi anemia or dyskeratosis congenita - may be misdiagnosed as having CAMT when they lack typical disease features of these syndromes or have only mild symptoms. This article reviews scientific and clinical aspects of the various disorders associated with the term "CAMT" with a main focus on the disease caused by mutations in the MPL gene.
Thrombocytopenia-absent radius (TAR) syndrome due to compound inheritance for a 1q21.1 microdeletion and a low-frequency noncoding RBM8A SNP: a new familial case.
Thrombocytopenia-absent radius syndrome (TAR; MIM 274000) is a rare autosomal recessive disorder combining specific skeletal abnormalities with a reduced platelet count. TAR syndrome has been associated with the compound inheritance of an interstitial microdeletion in 1q21.1 and a low frequency noncoding RBM8A SNP. Here, we report on a patient with scapulo-humeral hypoplasia, bilateral radio-ulnar agenesis with intact thumbs, bilateral proximal positioning of the first metacarpal, bilateral fifth finger clinodactyly, bilateral radial deviation of the hands, and thrombocytopenia. Molecular studies showed compound heterozygosity for the 1q21.1 microdeletion and the RBM8A rs139428292 variant in hemizygous state, inherited from the father and the mother, respectively. A second aborted fetus presented TAR features and 1q21.1 microdeletion. The complex inheritance pattern resulted in reduced expression of Y14, the protein encoded by RBM8A, and a component of the core exon-junction complex (EJC) in platelets. Further studies are needed to explain how Y14 insufficiency and subsequent defects of the EJC could cause the skeletal, haematological and additional features of TAR syndrome. In this study, we discuss other factors that could influence the overall phenotype of patients affected by TAR syndrome. In this study, we discuss other factors that could influence the overall phenotype of patients affected by TAR syndrome.
Publicações recentes
Proximal radio-ulnar synostosis with bone marrow failure syndrome in an infant without a HOXA11 mutation.
Inherited thrombocytopenias: toward a molecular understanding of disorders of platelet production.
"A new association of mental retardation, short stature, unusual face, radio-ulnar synostosis and retinal pigment abnormalities": Cohen syndrome with thrombocytopenia.
Congenital thrombocytopenia and radio-ulnar synostosis: a new familial syndrome.
📚 EuropePMCmostrando 3
Reduced-intensity conditioning is effective for allogeneic hematopoietic stem cell transplantation in infants with MECOM-associated syndrome.
International journal of hematologyCongenital amegakaryocytic thrombocytopenia - Not a single disease.
Best practice & research. Clinical haematologyThrombocytopenia-absent radius (TAR) syndrome due to compound inheritance for a 1q21.1 microdeletion and a low-frequency noncoding RBM8A SNP: a new familial case.
Molecular cytogeneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de sinostose radio-cubital-trombocitopenia amegacariocítica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de sinostose radio-cubital-trombocitopenia amegacariocítica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Reduced-intensity conditioning is effective for allogeneic hematopoietic stem cell transplantation in infants with MECOM-associated syndrome.
- Congenital amegakaryocytic thrombocytopenia - Not a single disease.
- Thrombocytopenia-absent radius (TAR) syndrome due to compound inheritance for a 1q21.1 microdeletion and a low-frequency noncoding RBM8A SNP: a new familial case.
- Proximal radio-ulnar synostosis with bone marrow failure syndrome in an infant without a HOXA11 mutation.
- Inherited thrombocytopenias: toward a molecular understanding of disorders of platelet production.
- "A new association of mental retardation, short stature, unusual face, radio-ulnar synostosis and retinal pigment abnormalities": Cohen syndrome with thrombocytopenia.
- Congenital thrombocytopenia and radio-ulnar synostosis: a new familial syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:71289(Orphanet)
- MONDO:0011555(MONDO)
- GARD:16687(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55783414(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
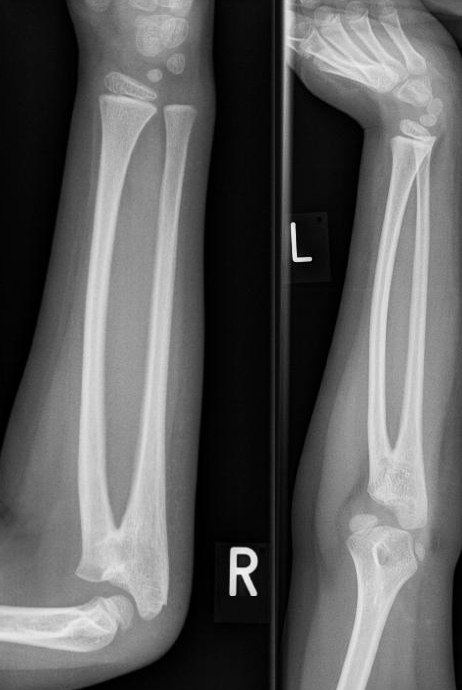
Síndrome de sinostose radio-cubital-trombocitopenia amegacariocítica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub