A síndrome de Stickler é uma vitreorretinopatia hereditária caracterizada pela associação de sinais oculares com formas mais ou menos completas da sequência de Pierre-Robin, distúrbios ósseos e surdez neurossensorial (10% dos casos). A síndrome de Stickler tipo 2 é causada por mutações no gene COL11A1 (1p21).
Introdução
O que você precisa saber de cara
A síndrome de Stickler é uma vitreorretinopatia hereditária caracterizada pela associação de sinais oculares com formas mais ou menos completas da sequência de Pierre-Robin, distúrbios ósseos e surdez neurossensorial (10% dos casos). A síndrome de Stickler tipo 2 é causada por mutações no gene COL11A1 (1p21).
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 12 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 39 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Curadoria gene-doença
fontes oficiaisMay play an important role in fibrillogenesis by controlling lateral growth of collagen II fibrils
Secreted, extracellular space, extracellular matrix
Stickler syndrome 2
An autosomal dominant form of Stickler syndrome, an inherited disorder that associates ocular signs with more or less complete forms of Pierre Robin sequence, bone disorders and sensorineural deafness. Ocular disorders may include juvenile cataract, myopia, strabismus, vitreoretinal or chorioretinal degeneration, retinal detachment, and chronic uveitis. Pierre Robin sequence includes an opening in the roof of the mouth (a cleft palate), a large tongue (macroglossia), and a small lower jaw (micrognathia). Bones are affected by slight platyspondylisis and large, often defective epiphyses. Juvenile joint laxity is followed by early signs of arthrosis. The degree of hearing loss varies among affected individuals and may become more severe over time. Syndrome expressivity is variable.
Variantes genéticas (ClinVar)
747 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 272 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
7 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de Stickler tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
An Unusual Retinal Presentation of a Novel COL11A1 Mutation: A Case Report.
Stickler syndrome is a rare collagenopathy, caused by mutations in various genes coding for fibrillar collagens II, IX, and XI. The disorder can be subdivided into different groups, depending on the genes affected and clinical features found in patients. Ocular symptoms, such as high myopia, retinal detachments, or anomalies in the vitreous, are present in most forms of Stickler syndrome. In this case report, we present a patient with an unusual retinal phenotype. Subject of this case report is a 33-year-old woman, who was examined at the Department of Ophthalmology at Medical University of Graz. A thorough ophthalmological examination was conducted, detailed medical and family history acquired, and genetic testing performed. Best corrected visual acuity was 20/20 on both eyes; however, impaired binocular vision associated with intermittent exotropia was found. Furthermore, dilated fundoscopy showed an unusual, hypopigmented spotted retinal phenotype. Fundus autofluorescence showed multiple hyperfluorescent spots corresponding with the spotted retinal appearance. Genetic testing revealed a novel variant in the gene COL11A1. No other ocular abnormalities which are associated with COL11A1 were found. Several subtypes of Stickler syndrome have been reported in medical literature, greatly varying in clinical manifestations. Many different mutations in the gene COL11A1 have been discovered and are typically associated with Stickler syndrome type 2. To our best knowledge, this is the first report of a patient with a mutation in the COL11A1 gene presenting with a hypopigmented spotted retina.
Genetic Variants in Rare Ophthalmological Syndromes: Novel COL11A1 Splice Site Variant in a Brazilian Family with Stickler Syndrome Type 2.
To genetically investigate a family with a clinical and ophthalmological phenotype suggestive of Stickler syndrome (STL) and the association of molecular findings with ophthalmological, clinical characteristics, and family history. Specialized ophthalmologic evaluation using diagnostic methods such as fundoscopy, slit-lamp photography, and ocular biometry to describe the phenotypic characteristics of the index patient and family members. Genetic evaluation with genome sequencing (WGS) is to describe the genotypic characteristics of the proband and confirmation of the finding by family-specific variant sequencing (NGS). Genetic evaluation demonstrated the presence of a novel pathogenic variant NM_001854.4: C.3168+1G>A (chr1:102961865-102961865), in the intronic region that follows exon 41 of the COL11A1 in the three affected members of the family. This variant is located 1 base from the natural splicing region of this exon and consequently can impact intron removal and protein formation. As for the ophthalmological findings, the presence of low visual acuity, high axial myopia, and pathological vitreous gel with a "beaded" appearance was observed in the index case. Further clinical examination showed that other family members also had vitreous and retinal degeneration. A heterozygous novel pathogenic variant in COL11A1 was identified by complete genome sequencing in a Brazilian family with STL. Ocular examination findings photographically presented confirm the characteristic features of STL type 2.
Microphthalmia and congenital cataract in two patients with Stickler syndrome type II: a case report.
Stickler syndrome (STL) is a collagenopathy caused by pathogenic variants in collagen-coding genes, mainly COL2A1 or COL11A1 associated with Stickler syndrome type 1 (STL1) or type 2 (STL2), respectively. Affected individuals manifest ocular, auditory, articular, and craniofacial findings in varying degrees. Previous literature and case reports describe high variability in clinical findings for patients with STL. With this case report, we broaden the clinical spectrum of the phenotype. Case report on two members of a family (mother and son) including clinical examination and genetic testing using targeted trio whole exome sequencing (trio-WES). A boy and his mother presented with microphthalmia, congenital cataract, ptosis, and moderate-to-severe sensorineural hearing loss. Trio-WES found a novel heterozygote missense variant, c.4526A>G; p(Gln1509Arg) in COL11A1 in both affected individuals. We report a previously undescribed phenotype associated with a COL11A1-variant in a mother and son, expanding the spectrum for phenotype-genotype correlation in STL2, presenting with microphthalmia, congenital cataract, and ptosis not normally associated with Stickler syndrome.
The Incidence of Velopharyngeal Insufficiency in Stickler Syndrome.
Stickler Syndrome (SS) is an inherited collagenopathy characterized by heterogenous orofacial, ocular, auditory, and skeletal abnormalities. The orofacial manifestations are variable and some patients present with cleft palate and velopharyngeal insufficiency (VPI). The incidence of VPI in SS is poorly studied and no studies have compared the incidence of VPI between Type I (COL2A1) and Type II (COL11A1) SS. The objective of this study is to compare the incidence of VPI between SS subtypes and discuss the surgical techniques used to treat them. Single-institution, retrospective chart review. Tertiary pediatric hospital. Forty-three children were diagnosed with SS between January 2003 and December 2018. Genetic testing results, genetics notes, craniofacial clinic notes, and operative reports were reviewed. Patients without genetic testing or craniofacial/otolaryngologic evaluation were excluded. Thirty-one patients met criteria and were included. Primary outcome was VPI incidence. There were 18 patients with Type I SS and 13 with Type II SS. Five (16%) patients had VPI, 2 (11%) with Type I SS compared to 3 (23%) with Type II SS (P > .05). All patients with VPI underwent surgery with either sphincter pharyngoplasty (3) or pharyngeal flap (2). Two patients with Type II SS underwent revision sphincter pharyngoplasty, with one conversion to pharyngeal flap. VPI is common for patients with SS. In this study, there was no significant difference in the incidence of VPI between SS subtypes. Future studies are needed to confirm these findings, which could be important for patient counseling and treatment planning.
Multiocular defect in the Old English Sheepdog: A canine form of Stickler syndrome type II associated with a missense variant in the collagen-type gene COL11A1.
Multiocular defect has been described in different canine breeds, including the Old English Sheepdog. Affected dogs typically present with multiple and various ocular abnormalities. We carried out whole genome sequencing on an Old English Sheepdog that had been diagnosed with hereditary cataracts at the age of five and then referred to a board-certified veterinary ophthalmologist due to owner-reported visual deterioration. An ophthalmic assessment revealed that there was bilateral vitreal degeneration, macrophthalmos, and spherophakia in addition to cataracts. Follow-up consultations revealed cataract progression, retinal detachment, uveitis and secondary glaucoma. Whole genome sequence filtered variants private to the case, shared with another Old English Sheepdog genome and predicted to be deleterious were genotyped in an initial cohort of six Old English Sheepdogs (three affected by multiocular defect and three control dogs without evidence of inherited eye disease). Only one of the twenty-two variants segregated correctly with multiocular defect. The variant is a single nucleotide substitution, located in the collagen-type gene COL11A1, c.1775T>C, that causes an amino acid change, p.Phe1592Ser. Genotyping of an additional 14 Old English Sheepdogs affected by multiocular defect revealed a dominant mode of inheritance with four cases heterozygous for the variant. Further genotyping of hereditary cataract-affected Old English Sheepdogs revealed segregation of the variant in eight out of nine dogs. In humans, variants in the COL11A1 gene are associated with Stickler syndrome type II, also dominantly inherited.
Publicações recentes
Genetic Variants in Rare Ophthalmological Syndromes: Novel COL11A1 Splice Site Variant in a Brazilian Family with Stickler Syndrome Type 2.
An Unusual Retinal Presentation of a Novel COL11A1 Mutation: A Case Report.
Exon-Trapping Assay Improves Clinical Interpretation of COL11A1 and COL11A2 Intronic Variants in Stickler Syndrome Type 2 and Otospondylomegaepiphyseal Dysplasia.
Homozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
Marshall and stickler syndrome in one family.
📚 EuropePMC355 artigos no totalmostrando 12
Genetic Variants in Rare Ophthalmological Syndromes: Novel COL11A1 Splice Site Variant in a Brazilian Family with Stickler Syndrome Type 2.
Journal of current ophthalmologyAn Unusual Retinal Presentation of a Novel COL11A1 Mutation: A Case Report.
Case reports in ophthalmologyMicrophthalmia and congenital cataract in two patients with Stickler syndrome type II: a case report.
Ophthalmic geneticsMultiocular defect in the Old English Sheepdog: A canine form of Stickler syndrome type II associated with a missense variant in the collagen-type gene COL11A1.
PloS oneThe Incidence of Velopharyngeal Insufficiency in Stickler Syndrome.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationExon-Trapping Assay Improves Clinical Interpretation of COL11A1 and COL11A2 Intronic Variants in Stickler Syndrome Type 2 and Otospondylomegaepiphyseal Dysplasia.
GenesOcular complications and prophylactic strategies in Stickler syndrome: a systematic literature review.
Ophthalmic geneticsHomozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
American journal of medical genetics. Part AMarshall and stickler syndrome in one family.
Ceska a slovenska oftalmologie : casopis Ceske oftalmologicke spolecnosti a Slovenske oftalmologicke spolecnostiA novel dominant COL11A1 mutation in a child with Stickler syndrome type II is associated with recurrent fractures.
Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USAA mild form of Stickler syndrome type II caused by mosaicism of COL11A1.
European journal of medical geneticsCephalometrics in Stickler syndrome: Objectification of the typical facial appearance.
Journal of cranio-maxillo-facial surgery : official publication of the European Association for Cranio-Maxillo-Facial SurgeryAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de Stickler tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de Stickler tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- An Unusual Retinal Presentation of a Novel COL11A1 Mutation: A Case Report.
- Genetic Variants in Rare Ophthalmological Syndromes: Novel COL11A1 Splice Site Variant in a Brazilian Family with Stickler Syndrome Type 2.
- Microphthalmia and congenital cataract in two patients with Stickler syndrome type II: a case report.
- The Incidence of Velopharyngeal Insufficiency in Stickler Syndrome.The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial Association· 2024· PMID 36443936mais citado
- Multiocular defect in the Old English Sheepdog: A canine form of Stickler syndrome type II associated with a missense variant in the collagen-type gene COL11A1.
- Exon-Trapping Assay Improves Clinical Interpretation of COL11A1 and COL11A2 Intronic Variants in Stickler Syndrome Type 2 and Otospondylomegaepiphyseal Dysplasia.
- Homozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2.
- Marshall and stickler syndrome in one family.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:90654(Orphanet)
- OMIM OMIM:604841(OMIM)
- MONDO:0011493(MONDO)
- GARD:5020(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55345775(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
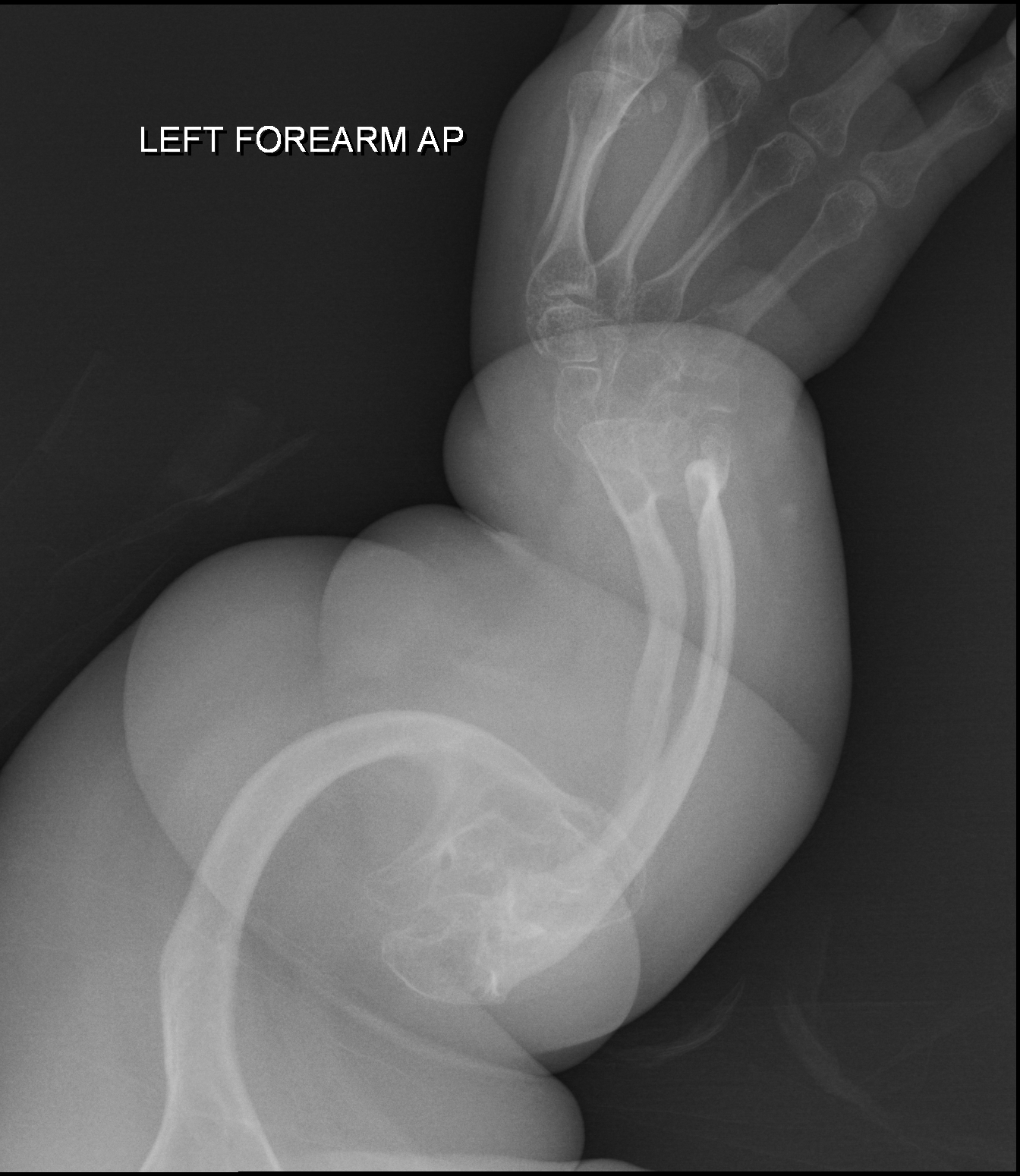
Síndrome de Stickler tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov