Erro congênito da formação de colágeno da cartilagem caracterizado por perda auditiva neurossensorial, epífises aumentadas, displasia esquelética com membros desproporcionalmente curtos, anomalias do corpo vertebral e uma fácies característica.
Introdução
O que você precisa saber de cara
Erro congênito da formação de colágeno da cartilagem caracterizado por perda auditiva neurossensorial, epífises aumentadas, displasia esquelética com membros desproporcionalmente curtos, anomalias do corpo vertebral e uma fácies característica.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 19 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 68 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisMay play an important role in fibrillogenesis by controlling lateral growth of collagen II fibrils
Secreted, extracellular space, extracellular matrix
Otospondylomegaepiphyseal dysplasia, autosomal dominant
An autosomal dominant form of otospondylomegaepiphyseal dysplasia, a disorder characterized by sensorineural deafness, enlarged epiphyses, mild platyspondyly, and disproportionate shortness of the limbs. Total body length is normal. Typical facial features are mid-face hypoplasia, short upturned nose and depressed nasal bridge. Most patients have Pierre Robin sequence including an opening in the roof of the mouth (cleft palate) and a small lower jaw (micrognathia). Ocular symptoms are absent. Some patients have early-onset osteoarthritis.
Type II collagen is specific for cartilaginous tissues. It is essential for the normal embryonic development of the skeleton, for linear growth and for the ability of cartilage to resist compressive forces
Secreted, extracellular space, extracellular matrix
Spondyloepiphyseal dysplasia congenital type
Disorder characterized by disproportionate short stature and pleiotropic involvement of the skeletal and ocular systems.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
2.070 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
13 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Displasia oto-espondilo-megaepifisária
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Case report: Autosomal recessive type 3 Stickler syndrome caused by compound heterozygous mutations in COL11A2.
Background: Stickler syndrome (SS) is a group of hereditary collagenopathies caused by a variety of collagen and non-collagen genes. Affected patients have characteristic manifestations involving ophthalmic, articular, craniofacial and auditory disorders. SS is classified into several subtypes according to clinical and molecular features. Type 3 SS is an ultra-rare disease, known as non-ocular SS or otospondylomegaepiphyseal dysplasia (OSMED) with only a few pathogenic COL11A2 variants reported to date. Case presentation: A 29-year-old Chinese male was referred to our hospital for hearing loss and multiple joint pain. He presented a phenotype highly suggestive of OSMED, including progressive sensorineural deafness, spondyloepiphyseal dysplasia with large epiphyses, platyspondyly, degenerative osteoarthritis, and sunken nasal bridge. We detected compound heterozygous mutations in COL11A2, both of which were predicted to be splicing mutations. One is synonymous mutation c.3774C>T (p.Gly1258Gly) supposed to be a splice site mutation, the other is a novel intron mutation c.4750 + 5 G>A, which is a highly conservative site across several species. We also present a review of the current known pathogenic mutation spectrum of COL11A2 in patients with type 3 SS. Conclusion: Both synonymous extonic and intronic variants are easily overlooked by whole-exome sequencing. For patients with clinical manifestations suspected of SS syndrome, next-generation whole-genome sequencing is necessary for precision diagnosis and genetic counseling.
Novel COL11A2 Pathogenic Variants in a Child with Autosomal Recessive Otospondylomegaepiphyseal Dysplasia: A Review of the Literature.
Otospondylomegaepiphyseal dysplasia (OSMED) is an inherited autosomal dominant and recessive skeletal dysplasia caused by both heterozygous and homozygous pathogenic variants in COL11A2 encoding the α2(XI) collagen chains, a part of type XI collagen. Here, we describe a 2-year-old girl presenting from birth with a phenotype suggestive of OSMED. On whole exome sequence analysis of the family via commercially available methods, we detected two novel heterozygous pathogenic variants in the proband. In addition, we reviewed the phenotype of autosomal recessive OSMED cases with COL11A2 pathogenic variants reported to date and quantitatively highlighted the phenotypic spectrum.
Novel mutations confirm that COL11A2 is responsible for autosomal recessive non-syndromic hearing loss DFNB53.
Hearing loss (HL) is a major public health issue. It is clinically and genetically heterogeneous.The identification of the causal mutation is important for early diagnosis, clinical follow-up, and genetic counseling. HL due to mutations in COL11A2, encoding collagen type XI alpha-2, can be non-syndromic autosomal-dominant or autosomal-recessive, and also syndromic as in Otospondylomegaepiphyseal Dysplasia, Stickler syndrome type III, and Weissenbacher-Zweymuller syndrome. However, thus far only one mutation co-segregating with autosomal recessive non-syndromic hearing loss (ARNSHL) in a single family has been reported. In this study, whole exome sequencing of two consanguineous families with ARNSHL from Tunisia and Turkey revealed two novel causative COL11A2 mutations, c.109G > T (p.Ala37Ser) and c.2662C > A (p.Pro888Thr). The variants identified co-segregated with deafness in both families. All homozygous individuals in those families had early onset profound hearing loss across all frequencies without syndromic findings. The variants are predicted to be damaging the protein function. The p.Pro888Thr mutation affects a -Gly-X-Y- triplet repeat motif. The novel p.Ala37Ser is the first missense mutation located in the NC4 domain of the COL11A2 protein. Structural model suggests that this mutation will likely obliterate, or at least partially compromise, the ability of NC4 domain to interact with its cognate ligands. In conclusion, we confirm that COL11A2 mutations cause ARNSHL and broaden the mutation spectrum that may shed new light on genotype-phenotype correlation for the associated phenotypes and clinical follow-up.
Publicações recentes
Lysosomal storage disorders in nonimmune hydrops fetalis diagnosed by exome sequencing.
📖 RevisãoDisease characteristics and clinical outcome over two decades from the Swiss pulmonary hypertension registry.
📚 EuropePMC1 artigos no totalmostrando 3
Case report: Autosomal recessive type 3 Stickler syndrome caused by compound heterozygous mutations in COL11A2.
Frontiers in geneticsNovel COL11A2 Pathogenic Variants in a Child with Autosomal Recessive Otospondylomegaepiphyseal Dysplasia: A Review of the Literature.
Journal of pediatric geneticsNovel mutations confirm that COL11A2 is responsible for autosomal recessive non-syndromic hearing loss DFNB53.
Molecular genetics and genomics : MGGAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Displasia oto-espondilo-megaepifisária.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Displasia oto-espondilo-megaepifisária
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Case report: Autosomal recessive type 3 Stickler syndrome caused by compound heterozygous mutations in COL11A2.
- Novel COL11A2 Pathogenic Variants in a Child with Autosomal Recessive Otospondylomegaepiphyseal Dysplasia: A Review of the Literature.
- Novel mutations confirm that COL11A2 is responsible for autosomal recessive non-syndromic hearing loss DFNB53.
- Lysosomal storage disorders in nonimmune hydrops fetalis diagnosed by exome sequencing.
- Disease characteristics and clinical outcome over two decades from the Swiss pulmonary hypertension registry.
- Correction: Phenotate: crowdsourcing phenotype annotations as exercises in undergraduate classes.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1427(Orphanet)
- MONDO:0008975(MONDO)
- GARD:4130(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7109017(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
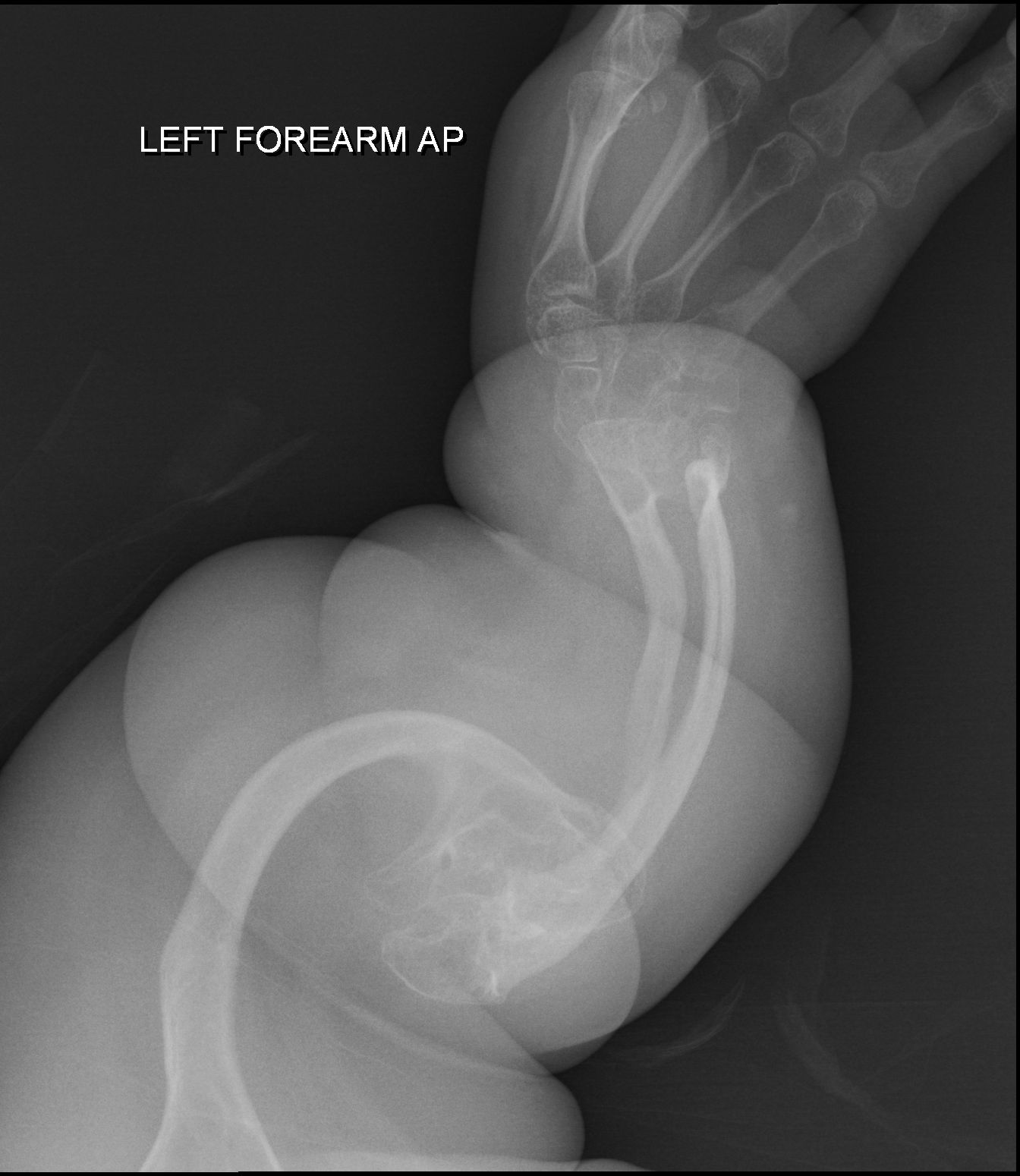
Displasia oto-espondilo-megaepifisária
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA