A síndrome de Desbuquois (DBQD) é uma osteocondrodisplasia caracterizada por nanismo micromélico grave, dismorfismo facial, frouxidão articular com múltiplas luxações, anormalidades vertebrais e metafisárias e ossificação carpotarsal avançada. Duas formas foram distinguidas com base na presença (tipo 1) ou na ausência (tipo 2) de anomalias características das mãos. Uma forma variante de DBQD, variante Kim, também foi descrita e é caracterizada por baixa estatura e anomalias articulares, faciais menores e anomalias significativas nas mãos.
Introdução
O que você precisa saber de cara
A síndrome de Desbuquois (DBQD) é uma osteocondrodisplasia caracterizada por nanismo micromélico grave, dismorfismo facial, frouxidão articular com múltiplas luxações, anormalidades vertebrais e metafisárias e ossificação carpotarsal avançada. Duas formas foram distinguidas com base na presença (tipo 1) ou na ausência (tipo 2) de anomalias características das mãos. Uma forma variante de DBQD, variante Kim, também foi descrita e é caracterizada por baixa estatura e anomalias articulares, faciais menores e anomalias significativas nas mãos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 42 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 113 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Transfers 1,4-N-acetylgalactosamine (GalNAc) from UDP-GalNAc to the non-reducing end of glucuronic acid (GlcUA). Required for addition of the first GalNAc to the core tetrasaccharide linker and for elongation of chondroitin chains. Important role in chondroitin chain biosynthesis in cartilage formation and subsequent endochondral ossification (PubMed:11788602, PubMed:12163485, PubMed:12446672, PubMed:17145758, PubMed:31705726). Moreover, is involved in the metabolism of aggrecan (By similarity)
Golgi apparatus, Golgi stack membrane
Skeletal dysplasia, mild, with joint laxity and advanced bone age
An autosomal recessive disorder characterized by skeletal dysplasia, short stature, short long bones, advanced bone age, joint laxity, and facial dysmorphism.
Catalyzes the first step in the biosynthesis of chondroitin sulfate and dermatan sulfate proteoglycans, such as DCN. Transfers D-xylose from UDP-D-xylose to specific serine residues of the core protein (PubMed:15461586, PubMed:17189265, PubMed:23982343, PubMed:24581741). Required for normal embryonic and postnatal skeleton development, especially of the long bones (PubMed:23982343, PubMed:24581741). Required for normal maturation of chondrocytes during bone development, and normal onset of ossif
Golgi apparatus membraneSecreted
Desbuquois dysplasia 2
A chondrodysplasia characterized by severe prenatal and postnatal growth retardation (less than -5 SD), joint laxity, short extremities, progressive scoliosis, round face, midface hypoplasia, prominent bulging eyes. The main radiologic features are short long bones with metaphyseal splay, a 'Swedish key' appearance of the proximal femur (exaggerated trochanter), and advance carpal and tarsal bone age. Two forms of Desbuquois dysplasia are distinguished on the basis of the presence or absence of characteristic hand anomalies: an extra ossification center distal to the second metacarpal, delta phalanx, bifid distal thumb phalanx, and phalangeal dislocations.
Calcium-dependent nucleotidase with a preference for UDP. The order of activity with different substrates is UDP > GDP > UTP > GTP. Has very low activity towards ADP and even lower activity towards ATP. Does not hydrolyze AMP and GMP (PubMed:12234496, PubMed:15006348, PubMed:15248776, PubMed:16835225). Involved in proteoglycan synthesis (PubMed:22539336)
Endoplasmic reticulum membraneGolgi apparatus, Golgi stack membraneCell membrane
Desbuquois dysplasia 1
A chondrodysplasia characterized by severe prenatal and postnatal growth retardation (less than -5 SD), joint laxity, short extremities, progressive scoliosis, round face, midface hypoplasia, prominent bulging eyes. The main radiologic features are short long bones with metaphyseal splay, a 'Swedish key' appearance of the proximal femur (exaggerated trochanter), and advance carpal and tarsal bone age. Two forms of Desbuquois dysplasia are distinguished on the basis of the presence or absence of characteristic hand anomalies: an extra ossification center distal to the second metacarpal, delta phalanx, bifid distal thumb phalanx, and phalangeal dislocations.
Variantes genéticas (ClinVar)
293 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Desbuquois
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Prenatal diagnosis and management of desbuquois dysplasia type 1 due to CANT1 mutation: A case report.
To report a rare case of Desbuquois dysplasia Type 1 (DBQD1) diagnosed prenatally, caused by mutations in the CANT1 gene. DBQD1 is an autosomal recessive skeletal dysplasia with severe disproportionate dwarfism, joint laxity, and multiple skeletal anomalies. A 26-year-old woman, gravida 3, para 1, abortus 1, was referred due to short femur (FL) and humerus (HL) lengths on ultrasound. The patient and her husband are consanguineous. Ultrasound at our clinic revealed a hypoplastic thorax, severe limb anomalies, brachydactyly, overlapping fingers, clubfoot, and rocker-bottom feet, along with ventricular septal defects (VSDs). Genetic testing identified a homozygous pathogenic variant in the CANT1 gene (c.902_906dup, p.Ser303AlafsTer21). Both parents were heterozygous carriers. Following genetic counseling, the family opted for pregnancy termination. This case highlights the importance of comprehensive genetic testing and early, precise diagnosis for informed decision-making in managing rare skeletal dysplasias.
From Desbuquois Dysplasia to Multiple Epiphyseal Dysplasia: The Clinical Impact of a CANT1 Variant Across Five Unrelated Families.
Multiple epiphyseal dysplasia (MED) is a heterogeneous group of chondrodysplasia characterized by arthralgia, early onset osteoarthropathy, and the radiographic findings of small, flat, and irregular-shaped epiphyses. Some patients with MED have mild short stature as well. MED is genetically heterogeneous caused by pathogenic variants in COMP, MATN3, COL9A1, COL9A2, COL9A3, and SLC26A2. In 2017, pathogenic variants in CANT1, which are responsible for Desbuquois dysplasia, have also been reported in the genetic etiology of MED. To date, only three patients have been reported with CANT1-related MED. Herein, we present clinical and radiographic findings of six additional patients from five unrelated families, all sharing the same c.375G > C; p.(Trp125Cys) variant in CANT1 gene. These patients exhibited the features of multiple epiphyseal dysplasia, along with some similarities to Desbuquois dysplasia, thereby broadening the clinical spectrum of CANT1-related disorders.
Clinical and Genetic Insights into Desbuquois Dysplasia: Review of 111 Case Reports.
Skeletal disorders encompass a wide array of conditions, many of which are associated with short stature. Among these, Desbuquois dysplasia is a rare but severe condition characterized by profound dwarfism, distinct facial features, joint hypermobility with multiple dislocations, and unique vertebral and metaphyseal anomalies. Desbuquois dysplasia is inherited in an autosomal recessive manner, with both the DBQD1 (MIM 251450) and DBQD2 (MIM 615777) forms resulting from biallelic mutations. Specifically, DBQD1 is associated with homozygous or compound heterozygous mutations in the CANT1 gene, while DBQD2 can result from mutations in either the CANT1 or XYLT1 genes. This review synthesizes the findings of 111 published case reports, including 54 cases of DBQD1, 39 cases of DBQD2, and 14 cases of the Kim variant (DDKV). Patients in this cohort had a median birth weight of 2505 g, a median length of 40 cm, and a median occipitofrontal circumference of 33 cm. The review highlights the phenotypic variations across Desbuquois dysplasia subtypes, particularly in facial characteristics, joint dislocations, and bone deformities. Genetic analyses revealed a considerable diversity in mutations, with over 35% of cases involving missense mutations, primarily affecting the CANT1 gene. Additionally, approximately 60% of patients had a history of parental consanguinity, indicating a potential genetic predisposition in certain populations. The identified mutations included deletions, insertions, and nucleotide substitutions, many of which resulted in premature stop codons and the production of truncated, likely nonfunctional proteins. These findings underscore the genetic and clinical complexity of Desbuquois dysplasia, highlighting the importance of early diagnosis and the potential for personalized therapeutic approaches. Continued research is essential to uncover the underlying mechanisms of this disorder and improve outcomes for affected individuals through targeted treatments.
Cant1 Affects Cartilage Proteoglycan Properties: Aggrecan and Decorin Characterization in a Mouse Model of Desbuquois Dysplasia Type 1.
Desbuquois dysplasia type 1 (DBQD1) is a recessive chondrodysplasia caused by mutations in the CANT1 gene, encoding for the Golgi Calcium-Activated Nucleotidase 1 (CANT1). The enzyme hydrolyzes UDP, the by-product of glycosyltransferase reactions, but it might play other roles in different cell types. Using a Cant1 knock-out mouse, we demonstrated that CANT1 is crucial for glycosaminoglycan (GAG) synthesis; however, its impact on the biochemical properties of cartilage proteoglycans remains unknown. Thus, in this work, we characterized decorin and aggrecan from primary chondrocyte cultures and cartilage biopsies of mutant mice at post-natal day 4 by Western blots and further investigated their distribution in the cartilage extracellular matrix (ECM) by immunohistochemistry. We demonstrated that the GAG synthesis defect caused by CANT1 impairment led to the synthesis and secretion of proteoglycans with shorter GAG chains compared with wild-type animals. However, this alteration did not result in the synthesis and secretion of decorin and aggrecan in the unglycanated form. Interestingly, the defect was not cartilage-specific since also skin decorin showed a reduced hydrodynamic size. Finally, immunohistochemical studies in epiphyseal sections of mutant mice demonstrated that the proteoglycan structural defect moderately affected decorin distribution in the ECM.
Antenatal Phenotype of Desbuquois Dysplasia.
Desbuquois dysplasia (DBQD) is an uncommon, autosomal recessive disorder with multiple joint dislocations. It is caused by pathogenic variants in CANT1 (calcium-activated nucleotidase 1) [NM_001159773.2]. This study adds to the scant data of nine reported antenatal phenotypes of DBQD. The present paper describes two unrelated consanguineous families with antenatal features of lethal skeletal dysplasia. The defining radiological changes were identified in only one patient who presented in the late second and third trimesters. Solo exome sequencing was performed and two previously reported homozygous variants c.896C>T (p.Pro299Leu) in patient 1 and c.902_906dup (p.Ser303fs*20) in patient 2 were identified. This study highlights the fetal presentations in DBQD and adds to its phenotypic spectrum. A complete clinical workup, including fetal autopsy and radiographs is essential to confirm the diagnosis of lethal skeletal dysplasia. Molecular diagnosis remains the diagnostic modality to define the causative variant. A definitive diagnosis is essential to inform management and offer reproductive care.
Publicações recentes
Endoplasmic reticulum retention of xylosyltransferase 1 (XYLT1) mutants underlying Desbuquois dysplasia type II.
💬 OpiniãoPhenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features.
📖 RevisãoSynophyrs, curly eyelashes and Ptyrigium colli in a girl with Desbuquois dysplasia: a case report and review of the literature.
🥇 DiretrizDesbuquois syndrome in three sisters with significantly different lengths of survival.
📖 RevisãoUse of CobraPLA for airway management in a neonate with Desbuquois syndrome. Case report and anesthetic implications.
📚 EuropePMC11 artigos no totalmostrando 21
Prenatal diagnosis and management of desbuquois dysplasia type 1 due to CANT1 mutation: A case report.
Taiwanese journal of obstetrics & gynecologyFrom Desbuquois Dysplasia to Multiple Epiphyseal Dysplasia: The Clinical Impact of a CANT1 Variant Across Five Unrelated Families.
American journal of medical genetics. Part ACant1 Affects Cartilage Proteoglycan Properties: Aggrecan and Decorin Characterization in a Mouse Model of Desbuquois Dysplasia Type 1.
BiomoleculesClinical and Genetic Insights into Desbuquois Dysplasia: Review of 111 Case Reports.
International journal of molecular sciencesAntenatal Phenotype of Desbuquois Dysplasia.
Indian journal of pediatricsNovel compound heterozygous variants in XYLT1 gene caused Desbuquois dysplasia type 2 in an aborted fetus: a case report.
BMC pediatricsPhenotypic Characterization of Immortalized Chondrocytes from a Desbuquois Dysplasia Type 1 Mouse Model: A Tool for Studying Defects in Glycosaminoglycan Biosynthesis.
International journal of molecular sciencesDesbuquois dysplasia Kim variant: a rare case report syndrome.
Clinical dysmorphologyCANT1 deficiency in a mouse model of Desbuquois dysplasia impairs glycosaminoglycan synthesis and chondrocyte differentiation in growth plate cartilage.
FEBS open bioPosterior vertebral column resection for rigid proximal thoracic kyphoscoliosis with broken growing rods in a patient with Desbuquois dysplasia.
Spine deformity[Analysis of CANT1 gene variant in a girl with Desbuquois dysplasia type I].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsPrenatal diagnosis of Desbuquois dysplasia Type 1: Utilization of high-density SNP array to map homozygosity and identify the gene.
American journal of medical genetics. Part APrenatal diagnosis of Desbuquois dysplasia type 1 by whole exome sequencing before the occurrence of specific ultrasound signs.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansA novel homozygous variant in CANT1 in a patient with Kim-type Desbuquois dysplasia.
Clinical dysmorphologyA novel gene (FAM20B encoding glycosaminoglycan xylosylkinase) for neonatal short limb dysplasia resembling Desbuquois dysplasia.
Clinical geneticsCalcium activated nucleotidase 1 (CANT1) is critical for glycosaminoglycan biosynthesis in cartilage and endochondral ossification.
Matrix biology : journal of the International Society for Matrix BiologyEndoplasmic reticulum retention of xylosyltransferase 1 (XYLT1) mutants underlying Desbuquois dysplasia type II.
American journal of medical genetics. Part ADesbuquois dysplasia type II in a patient with a homozygous mutation in XYLT1 and new unusual findings.
American journal of medical genetics. Part AExome sequencing reveals two novel compound heterozygous XYLT1 mutations in a Polish patient with Desbuquois dysplasia type 2 and growth hormone deficiency.
Journal of human geneticsMutations in Biosynthetic Enzymes for the Protein Linker Region of Chondroitin/Dermatan/Heparan Sulfate Cause Skeletal and Skin Dysplasias.
BioMed research international[Two siblings with bilateral congenital knee dislocations: case report].
Acta chirurgiae orthopaedicae et traumatologiae CechoslovacaAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Desbuquois.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Desbuquois
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Prenatal diagnosis and management of desbuquois dysplasia type 1 due to CANT1 mutation: A case report.
- From Desbuquois Dysplasia to Multiple Epiphyseal Dysplasia: The Clinical Impact of a CANT1 Variant Across Five Unrelated Families.
- Clinical and Genetic Insights into Desbuquois Dysplasia: Review of 111 Case Reports.
- Cant1 Affects Cartilage Proteoglycan Properties: Aggrecan and Decorin Characterization in a Mouse Model of Desbuquois Dysplasia Type 1.
- Antenatal Phenotype of Desbuquois Dysplasia.
- Endoplasmic reticulum retention of xylosyltransferase 1 (XYLT1) mutants underlying Desbuquois dysplasia type II.
- Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features.
- Synophyrs, curly eyelashes and Ptyrigium colli in a girl with Desbuquois dysplasia: a case report and review of the literature.
- Desbuquois syndrome in three sisters with significantly different lengths of survival.
- Use of CobraPLA for airway management in a neonate with Desbuquois syndrome. Case report and anesthetic implications.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1425(Orphanet)
- MONDO:0015426(MONDO)
- GARD:1818(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q17122800(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
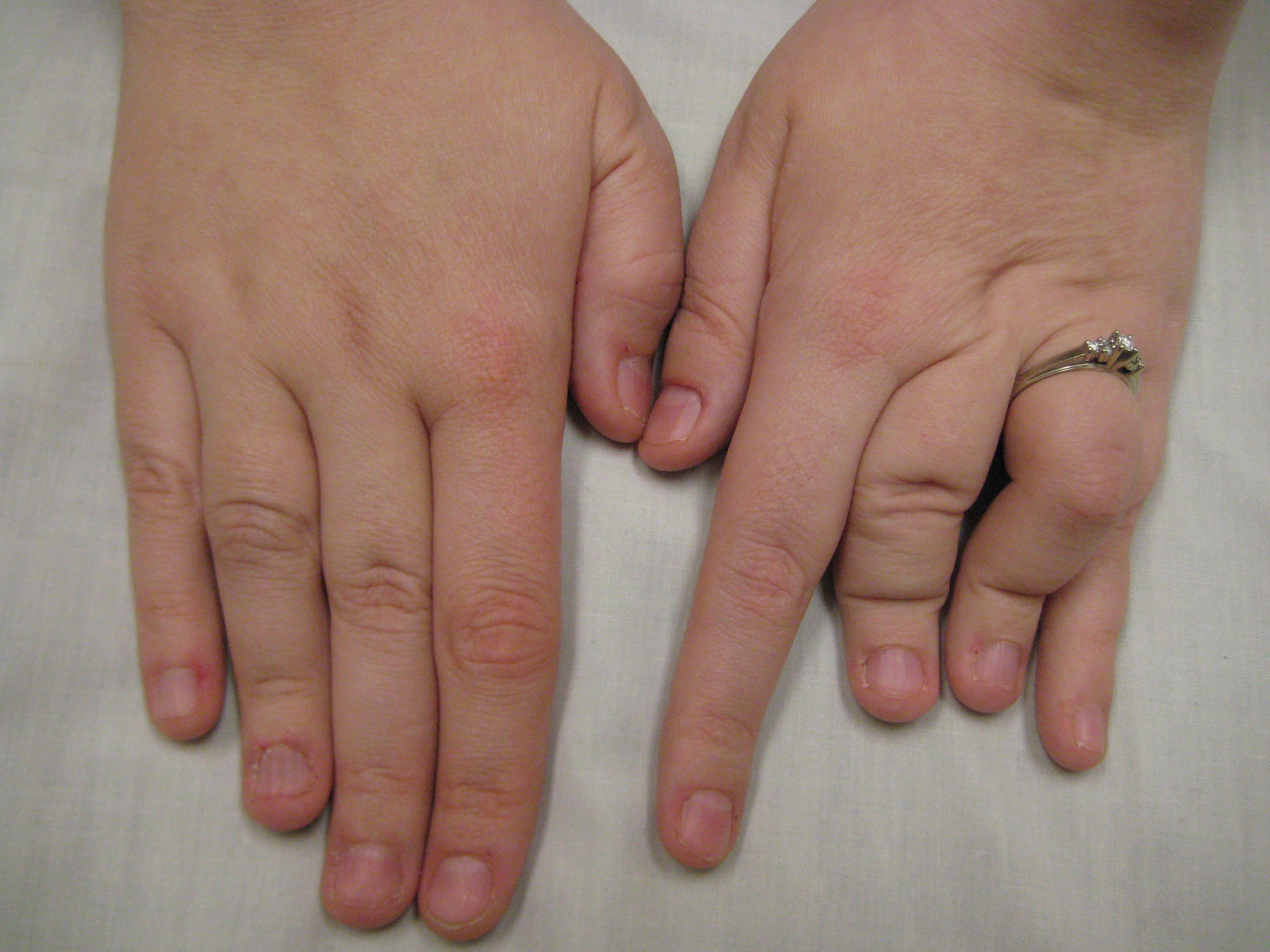
Síndrome Desbuquois
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata