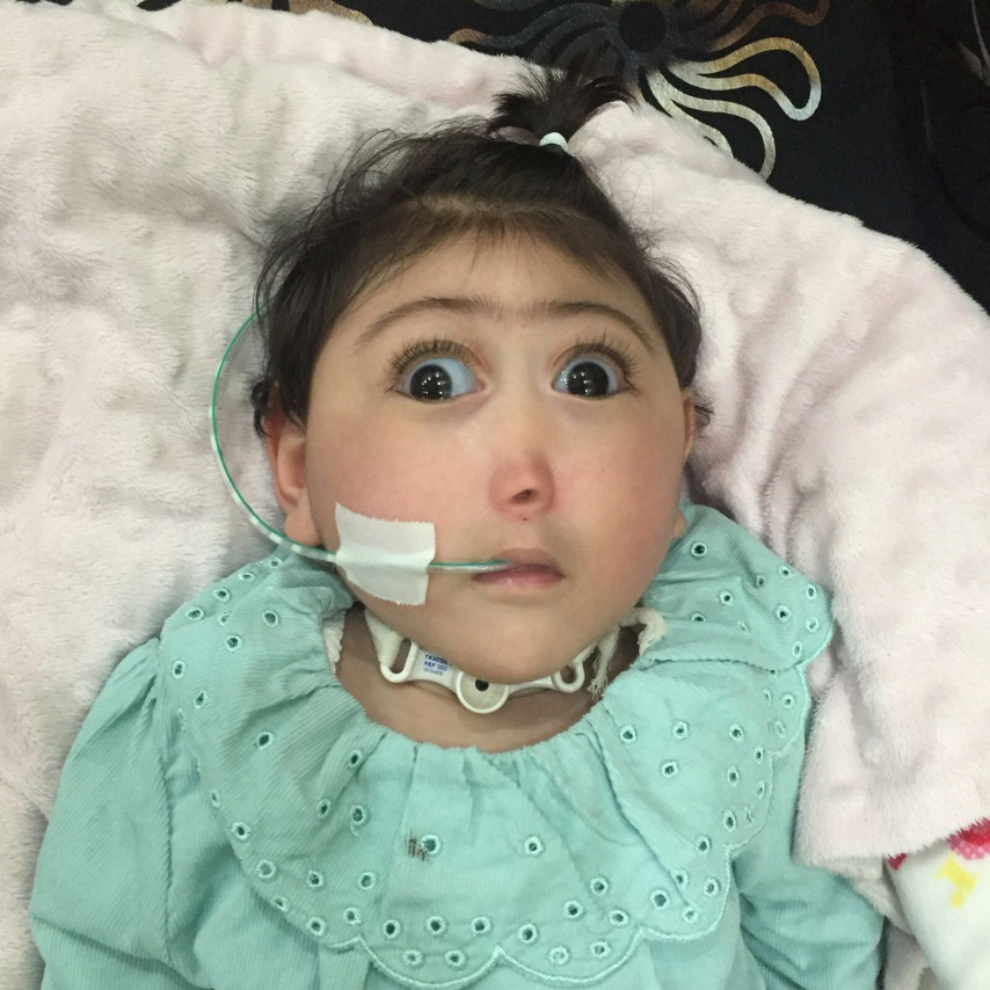
Síndrome Hartsfield é uma condição rara autossômica dominante caracterizada por orelhas de implantação baixa, atraso de crescimento, holoprosencefalia semilobar, microcefalia e diabetes insipidus. A agenesia do corpo caloso e deficiência de gonadotrofina também são comuns.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome Hartsfield é uma condição genética rara, com prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. Ela se manifesta ainda durante a gestação (período antenatal) e pode afetar múltiplos sistemas do corpo, incluindo o desenvolvimento do cérebro, do crânio, da face e dos membros.[1][4]
Sinais e sintomas
Os sinais e sintomas da Síndrome Hartsfield são variados e podem incluir: malformações cerebrais como holoprosencefalia (formas lobar e alobar), ausência ou subdesenvolvimento do corpo caloso (aplasia/hipoplasia do corpo caloso) e encefalocele. Na face, podem ocorrer fenda labial (não mediana) e/ou fenda palatina, hipertelorismo (aumento da distância entre os olhos) ou hipotelorismo (diminuição dessa distância), telecanto, fissuras palpebrais inclinadas para baixo, ptose (queda da pálpebra), microftalmia (olho pequeno), ponte nasal deprimida e orelhas com rotação posterior. O crânio pode apresentar craniossinostose (fechamento precoce das suturas cranianas). Nos membros, podem ser observadas mão fendida (ectrodactilia), sindactilia (dedos unidos), ausência ou subdesenvolvimento do rádio (aplasia/hipoplasia do rádio). Outros achados incluem retardo do crescimento intrauterino, hipospadia (abertura da uretra na face inferior do pênis), hipotonia neonatal (tônus muscular baixo ao nascer), atraso global do desenvolvimento, insuficiência respiratória e hipernatremia (nível elevado de sódio no sangue).[1][4]
Causas genéticas
A Síndrome Hartsfield é causada por alterações (variantes patogênicas) no gene FGFR1 (Fibroblast Growth Factor Receptor 1). Esse gene fornece instruções para a produção de uma proteína importante para o desenvolvimento embrionário, especialmente do cérebro, crânio e membros. A condição pode ser herdada de forma autossômica dominante (quando uma única cópia alterada do gene é suficiente para causar a doença) ou autossômica recessiva (quando duas cópias alteradas do gene são necessárias).[1][2][5]
Diagnóstico
O diagnóstico da Síndrome Hartsfield é baseado na avaliação clínica dos sinais e sintomas característicos e pode ser confirmado por testes genéticos. Os exames disponíveis incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína também pode ser realizada. Atualmente, existem 28 testes genéticos disponíveis e 468 variantes descritas no ClinVar para essa condição.[1][2][5]
Tratamento e manejo
Não há cura para a Síndrome Hartsfield, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O acompanhamento deve ser multidisciplinar, envolvendo especialistas como geneticistas, neurologistas, cirurgiões craniofaciais, ortopedistas, fonoaudiólogos e terapeutas ocupacionais. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para o atendimento, incluindo procedimentos como cariótipo, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES), dosagem de alfa-fetoproteína e atendimento em reabilitação para doenças raras.[1][2]
Prognóstico e qualidade de vida
O prognóstico da Síndrome Hartsfield varia amplamente, dependendo da gravidade das malformações, especialmente as cerebrais e respiratórias. O atraso global do desenvolvimento e as complicações respiratórias podem impactar significativamente a qualidade de vida. O acompanhamento médico regular e as intervenções de reabilitação são essenciais para otimizar o desenvolvimento e o bem-estar do paciente.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome Hartsfield é uma condição rara autossômica dominante caracterizada por orelhas de implantação baixa, atraso de crescimento, holoprosencefalia semilobar, microcefalia e diabetes insipidus. A agenesia do corpo caloso e deficiência de gonadotrofina também são comuns.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome Hartsfield é uma condição genética rara, com prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. Ela se manifesta ainda durante a gestação (período antenatal) e pode afetar múltiplos sistemas do corpo, incluindo o desenvolvimento do cérebro, do crânio, da face e dos membros.[1][4]
Sinais e sintomas
Os sinais e sintomas da Síndrome Hartsfield são variados e podem incluir: malformações cerebrais como holoprosencefalia (formas lobar e alobar), ausência ou subdesenvolvimento do corpo caloso (aplasia/hipoplasia do corpo caloso) e encefalocele. Na face, podem ocorrer fenda labial (não mediana) e/ou fenda palatina, hipertelorismo (aumento da distância entre os olhos) ou hipotelorismo (diminuição dessa distância), telecanto, fissuras palpebrais inclinadas para baixo, ptose (queda da pálpebra), microftalmia (olho pequeno), ponte nasal deprimida e orelhas com rotação posterior. O crânio pode apresentar craniossinostose (fechamento precoce das suturas cranianas). Nos membros, podem ser observadas mão fendida (ectrodactilia), sindactilia (dedos unidos), ausência ou subdesenvolvimento do rádio (aplasia/hipoplasia do rádio). Outros achados incluem retardo do crescimento intrauterino, hipospadia (abertura da uretra na face inferior do pênis), hipotonia neonatal (tônus muscular baixo ao nascer), atraso global do desenvolvimento, insuficiência respiratória e hipernatremia (nível elevado de sódio no sangue).[1][4]
Causas genéticas
A Síndrome Hartsfield é causada por alterações (variantes patogênicas) no gene FGFR1 (Fibroblast Growth Factor Receptor 1). Esse gene fornece instruções para a produção de uma proteína importante para o desenvolvimento embrionário, especialmente do cérebro, crânio e membros. A condição pode ser herdada de forma autossômica dominante (quando uma única cópia alterada do gene é suficiente para causar a doença) ou autossômica recessiva (quando duas cópias alteradas do gene são necessárias).[1][2][5]
Diagnóstico
O diagnóstico da Síndrome Hartsfield é baseado na avaliação clínica dos sinais e sintomas característicos e pode ser confirmado por testes genéticos. Os exames disponíveis incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína também pode ser realizada. Atualmente, existem 28 testes genéticos disponíveis e 468 variantes descritas no ClinVar para essa condição.[1][2][5]
Tratamento e manejo
Não há cura para a Síndrome Hartsfield, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O acompanhamento deve ser multidisciplinar, envolvendo especialistas como geneticistas, neurologistas, cirurgiões craniofaciais, ortopedistas, fonoaudiólogos e terapeutas ocupacionais. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para o atendimento, incluindo procedimentos como cariótipo, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES), dosagem de alfa-fetoproteína e atendimento em reabilitação para doenças raras.[1][2]
Prognóstico e qualidade de vida
O prognóstico da Síndrome Hartsfield varia amplamente, dependendo da gravidade das malformações, especialmente as cerebrais e respiratórias. O atraso global do desenvolvimento e as complicações respiratórias podem impactar significativamente a qualidade de vida. O acompanhamento médico regular e as intervenções de reabilitação são essenciais para otimizar o desenvolvimento e o bem-estar do paciente.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 41 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Curadoria gene-doença
fontes oficiaisTyrosine-protein kinase that acts as a cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of embryonic development, cell proliferation, differentiation and migration. Required for normal mesoderm patterning and correct axial organization during embryonic development, normal skeletogenesis and normal development of the gonadotropin-releasing hormone (GnRH) neuronal system. Phosphorylates PLCG1, FRS2, GAB1 and SHB. Ligand binding leads to the activati
Cell membraneNucleusCytoplasm, cytosolCytoplasmic vesicle
Pfeiffer syndrome
A syndrome characterized by the association of craniosynostosis, broad and deviated thumbs and big toes, and partial syndactyly of the fingers and toes. Three subtypes are known: mild autosomal dominant form (type 1); cloverleaf skull, elbow ankylosis, early death, sporadic (type 2); craniosynostosis, early demise, sporadic (type 3).
Variantes genéticas (ClinVar)
468 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
22 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Hartsfield
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Prenatal Diagnosis of Hartsfield Syndrome in the Fetus With Isolated Ectrodactyly Caused by a Novel Variant in FGFR1.
Molecular genetic testing was performed on a fetus with ectrodactyly of the right foot to clarify the pathogenic cause and provide evidence for prenatal counseling. Trio whole-exome sequencing (trio-WES) was performed on the fetus and his parents to identify the underlying genetic cause. Candidate variants were validated by Sanger sequencing, and their molecular effects were analyzed through minigene assays. Trio-WES identified a novel heterozygous variant (c.1977+1G>C) in FGFR1, which is consistent with FGFR1-related Hartsfield syndrome (HS; OMIM#615465). Sanger sequencing confirmed that this variant was de novo. The minigene assay revealed that all variants (c.1977+1G>C, c.1977+1G>A, and c.1977+1G>T) at the splice site generated two aberrant splicing events: (1) complete retention of intron 14, leading to a frameshift and premature termination codon; and (2) skipping of exon 14, causing an in-frame deletion of 41 amino acids. These events collectively impaired the function of the FGFR1 protein's tyrosine kinase domain. To our knowledge, prenatal reports of FGFR1-related HS remain extremely limited, and this is the first molecularly confirmed prenatal diagnosis of HS in China. The findings not only expand the mutational spectrum of HS but also provide genetic counseling and reproductive guidance for this family.
Malformation Pattern and Molecular Findings in the FGFR1-Related Hartsfield Syndrome Phenotype.
The Fibroblast Growth Factor Receptor 1 (FGFR1, MIM*136350) is a protein member of the fibroblast growth factor receptor (FGFR) family, with various biological functions, such as the normal development control. It contains an extracellular site for the ligand (three Ig-like domains, IgI, IgII, IgIII), a single transmembrane and a cytoplasmic protein tyrosine kinase (TK) domain. Variants in this gene have been associated with a wide spectrum of genetic disorders, including the clinical entity known as FGFR1-related Hartsfield or Hartsfield syndrome (HRTFDS, MIM#615465), which is an autosomal dominant or recessive disorder characterized by the clinical association of split-hand/foot malformation (SHFM) and holoprosencephaly (HPE). Dysmorphic facies, including cleft/lip palate, genitourinary anomalies, cardiovascular defects and intellectual disability/developmental delay (ID/DD) can also be a part of the clinical picture. The malformation phenotype of HRTFDS has been reviewed in 26 previously reported patients in terms of single congenital defects, mutational spectrum, impacted protein domains and inheritance. Molecular basis, clinical management, main differential diagnoses and genetic counseling were also illustrated. SHFM was identified in every patient. The other main associated features included craniofacial defects, skeletal malformation identified at radiography, genitourinary anomalies, HPE and cardiovascular disorders. FGFR1 causative variants mainly impact the TK domain and have a smaller impact on other protein sites (IgII, IgIII). This study extensively recapitulates the malformation phenotype associated with HRTFDS and the underlying molecular perturbations. A multidisciplinary clinical approach is fundamental, in which genetic counseling can have an important role. However, our results are partial and refer to a restricted number of patients, pointing out the necessity of other descriptions and similar research. Additional studies will expand clinical and molecular knowledge as well as further clarify the biological mechanisms.
Intrafamilial Phenotypic Variability of the FGFR1 p.Cys277Tyr Variant: A Case Report and Review of the Literature.
Split-hand/foot malformation (SHFM) is a rare congenital limb anomaly defined by the absence or hypoplasia of the central rays of the autopod. SHFM occurs as an isolated entity or part of genetic syndromes with several causative copy-number variations or monogenic alterations known to be involved in the disease pathomechanism. On the other hand, cleft lip/palate (CL/P) usually results from polygenic and environmental factors, with the complex interplay of both leading to this malformation. Pathogenic variants in FGFR1 have been linked to phenotypically distinct disorders, including Hartsfield syndrome, Kallmann syndrome, Jackson-Weiss syndrome, osteoglophonic dysplasia, and Pfeiffer syndrome. Although pathogenic variants in FGFR1 can contribute to syndromic SHFM or CL/P, their role in isolated SHFM or CL remains poorly described in the literature. We conducted targeted next-generation sequencing (NGS) in the proband with SHFM, followed by segregation analysis in the family members. In this study, we report an index patient presenting with isolated SHFM and his brother with CL and facial dysmorphism, as well as their father with isolated hyposmia. Targeted next-generation sequencing revealed a previously reported heterozygous missense pathogenic variant in FGFR1 (c.830G>A; p.Cys277Tyr) in both affected siblings and their hyposmic father. This study expands the phenotypic spectrum associated with FGFR1 pathogenic variants, emphasizing their involvement in non-syndromic SHFM and CL or isolated hyposmia. Our findings highlight the importance of considering FGFR1 in the molecular diagnosis of isolated SHFM or orofacial clefting, point to the high intrafamilial variability of FGFR1 pathogenic variants, and demonstrate the diagnostic value of targeted NGS in rare congenital malformations.
Novel Missense Variant in the FGFR1 Gene Associated With Prenatal Diagnosis of Hartsfield Syndrome.
Prenatal identification of a pathogenic maternal FGFR1 variant in two consecutive pregnancies with fetal forebrain malformations.
Holoprosencephaly (HPE) is the most common aberration of forebrain development, and it leads to a wide spectrum of developmental and craniofacial anomalies. HPE etiology is highly heterogeneous and includes both chromosomal abnormalities and single-gene defects. Here, we report an FGFR1 heterozygous variant detected by prenatal exome sequencing and inherited from the asymptomatic mother, in association with recurrent neurological abnormalities in the HPE spectrum in two consecutive pregnancies. Individuals with germline pathogenic variants in FGFR1 (MIM: 136350) show extensive phenotypic variability, which ranges from asymptomatic carriers to hypogonadotropic hypogonadism, arhinencephaly, Kallmann's syndrome with associated features such as cleft lip and palate, skeletal anomalies, isolated HPE, and Hartsfield syndrome. The presented case supports the role of exome sequencing in prenatal diagnosis when fetal midline structural anomalies are suggestive of a genetic etiology, as early as the first trimester of gestation. The profound heterogeneity of FGFR1 allelic disorders needs to be considered when planning prenatal screening even in asymptomatic carriers.
Publicações recentes
Malformation Pattern and Molecular Findings in the FGFR1-Related Hartsfield Syndrome Phenotype.
Novel Missense Variant in the FGFR1 Gene Associated With Prenatal Diagnosis of Hartsfield Syndrome.
Prenatal Diagnosis of Hartsfield Syndrome in the Fetus With Isolated Ectrodactyly Caused by a Novel Variant in FGFR1.
Intrafamilial Phenotypic Variability of the FGFR1 p.Cys277Tyr Variant: A Case Report and Review of the Literature.
Prenatal identification of a pathogenic maternal FGFR1 variant in two consecutive pregnancies with fetal forebrain malformations.
📚 EuropePMC18 artigos no totalmostrando 17
Malformation Pattern and Molecular Findings in the FGFR1-Related Hartsfield Syndrome Phenotype.
Medical sciences (Basel, Switzerland)Novel Missense Variant in the FGFR1 Gene Associated With Prenatal Diagnosis of Hartsfield Syndrome.
Prenatal diagnosisPrenatal Diagnosis of Hartsfield Syndrome in the Fetus With Isolated Ectrodactyly Caused by a Novel Variant in FGFR1.
American journal of medical genetics. Part AIntrafamilial Phenotypic Variability of the FGFR1 p.Cys277Tyr Variant: A Case Report and Review of the Literature.
GenesPrenatal identification of a pathogenic maternal FGFR1 variant in two consecutive pregnancies with fetal forebrain malformations.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansPrenatal diagnosis of Hartsfield syndrome with a novel genetic variant.
Prenatal diagnosisSolitary Median Maxillary Central Incisor in Hartsfield Syndrome: A Case Report.
International journal of clinical pediatric dentistryMosaicism in Hartsfield syndrome.
European journal of medical geneticsEndocrinological Features of Hartsfield Syndrome in an Adult Patient With a Novel Mutation of FGFR1.
Journal of the Endocrine SocietyNovel synonymous and missense variants in FGFR1 causing Hartsfield syndrome.
American journal of medical genetics. Part AA novel dominant-negative FGFR1 variant causes Hartsfield syndrome by deregulating RAS/ERK1/2 pathway.
European journal of human genetics : EJHGThe Use of Variant Maps to Explore Domain-Specific Mutations of FGFR1.
Journal of dental researchOtorhinolaryngologic manifestations of Hartsfield syndrome: Case series and review of literature.
International journal of pediatric otorhinolaryngologyNovel heterozygous mutation in the extracellular domain of FGFR1 associated with Hartsfield syndrome.
Human genome variationDetection of gonadal mosaicism in Hartsfield syndrome by next generation sequencing.
American journal of medical genetics. Part AHartsfield syndrome associated with a novel heterozygous missense mutation in FGFR1 and incorporating tumoral calcinosis.
American journal of medical genetics. Part ADominant-negative kinase domain mutations in FGFR1 can explain the clinical severity of Hartsfield syndrome.
Human molecular geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Hartsfield.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Hartsfield
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Prenatal Diagnosis of Hartsfield Syndrome in the Fetus With Isolated Ectrodactyly Caused by a Novel Variant in FGFR1.
- Malformation Pattern and Molecular Findings in the FGFR1-Related Hartsfield Syndrome Phenotype.
- Intrafamilial Phenotypic Variability of the FGFR1 p.Cys277Tyr Variant: A Case Report and Review of the Literature.
- Novel Missense Variant in the FGFR1 Gene Associated With Prenatal Diagnosis of Hartsfield Syndrome.
- Prenatal identification of a pathogenic maternal FGFR1 variant in two consecutive pregnancies with fetal forebrain malformations.The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstetricians· 2024· PMID 38679587mais citado
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2117(Orphanet)
- OMIM OMIM:615465(OMIM)
- MONDO:0014196(MONDO)
- GARD:2725(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55784727(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Hartsfield
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata