A síndrome de Meckel-Gruber é um distúrbio genético ciliopático raro e letal, caracterizado por displasia renal cística, malformações do sistema nervoso central, polidactilia (pós-axial), defeitos no desenvolvimento hepático e hipoplasia pulmonar devido ao oligodramnio. A síndrome de Meckel-Gruber recebe o nome de Johann Meckel e Georg Gruber. O prognóstico para bebês nascidos com a síndrome de Meckel-Gruber é reservado, sendo a maioria natimorta ou falecendo dentro de horas ou dias.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome Meckel-like NPHP3-relacionado é uma doença genética rara, com prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. Ela se manifesta já na infância e afeta múltiplos órgãos, principalmente os rins, o fígado e o sistema nervoso central. O nome "Meckel-like" indica que a condição se assemelha à Síndrome de Meckel, porém é causada por alterações em um gene específico, o NPHP3.[1][4]
Sinais e sintomas
Os sinais e sintomas da síndrome são variados e podem incluir: displasia renal (desenvolvimento anormal dos rins) ou displasia renal multicística, doença renal crônica estágio 5 (insuficiência renal avançada), múltiplos cistos glomerulares, morfologia anormal do parênquima hepático (tecido do fígado), proliferação dos ductos biliares, colestase (acúmulo de bile), cirrose biliar, hipertensão portal, hepatoesplenomegalia (aumento do fígado e baço), cistos pancreáticos, má rotação intestinal, hipoplasia pulmonar (pulmões subdesenvolvidos), oligodramnia ou polidrâmnio (quantidade anormal de líquido amniótico durante a gestação), malformação de Dandy-Walker (alteração na parte posterior do crânio), cisto do plexo coroide, hipertonia (aumento do tônus muscular), fontanelas grandes, testa alta, polidactilia pós-axial do pé (dedo extra no pé), hipertrofia ventricular direita e estenose da valva aórtica.[1][4]
Causas genéticas
A síndrome é causada por alterações (mutações) no gene NPHP3, que fornece instruções para a produção da proteína nefrocistina-3. A herança é autossômica recessiva, ou seja, a pessoa precisa herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença. Pais que são portadores de uma única cópia alterada geralmente não apresentam sintomas.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais e sintomas e confirmado por testes genéticos. Os exames disponíveis incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína também pode ser realizada como parte da investigação. Atualmente, há 49 testes genéticos disponíveis e 261 variantes descritas no ClinVar relacionadas a esta condição.[1][2][5]
Tratamento e manejo
O manejo da Síndrome Meckel-like NPHP3-relacionado é multidisciplinar e focado no tratamento dos sintomas e complicações. Não há cura específica. O acompanhamento pode incluir nefrologista (para doença renal), hepatologista (para problemas hepáticos), cardiologista (para alterações cardíacas) e geneticista. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para esta condição, incluindo atendimento em reabilitação para doenças raras. Não há medicamentos específicos aprovados para a síndrome.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas, especialmente o comprometimento renal e hepático. A doença renal crônica estágio 5 pode exigir diálise ou transplante renal. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para melhorar a qualidade de vida e o manejo das complicações.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A síndrome de Meckel-Gruber é um distúrbio genético ciliopático raro e letal, caracterizado por displasia renal cística, malformações do sistema nervoso central, polidactilia (pós-axial), defeitos no desenvolvimento hepático e hipoplasia pulmonar devido ao oligodramnio. A síndrome de Meckel-Gruber recebe o nome de Johann Meckel e Georg Gruber. O prognóstico para bebês nascidos com a síndrome de Meckel-Gruber é reservado, sendo a maioria natimorta ou falecendo dentro de horas ou dias.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome Meckel-like NPHP3-relacionado é uma doença genética rara, com prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. Ela se manifesta já na infância e afeta múltiplos órgãos, principalmente os rins, o fígado e o sistema nervoso central. O nome "Meckel-like" indica que a condição se assemelha à Síndrome de Meckel, porém é causada por alterações em um gene específico, o NPHP3.[1][4]
Sinais e sintomas
Os sinais e sintomas da síndrome são variados e podem incluir: displasia renal (desenvolvimento anormal dos rins) ou displasia renal multicística, doença renal crônica estágio 5 (insuficiência renal avançada), múltiplos cistos glomerulares, morfologia anormal do parênquima hepático (tecido do fígado), proliferação dos ductos biliares, colestase (acúmulo de bile), cirrose biliar, hipertensão portal, hepatoesplenomegalia (aumento do fígado e baço), cistos pancreáticos, má rotação intestinal, hipoplasia pulmonar (pulmões subdesenvolvidos), oligodramnia ou polidrâmnio (quantidade anormal de líquido amniótico durante a gestação), malformação de Dandy-Walker (alteração na parte posterior do crânio), cisto do plexo coroide, hipertonia (aumento do tônus muscular), fontanelas grandes, testa alta, polidactilia pós-axial do pé (dedo extra no pé), hipertrofia ventricular direita e estenose da valva aórtica.[1][4]
Causas genéticas
A síndrome é causada por alterações (mutações) no gene NPHP3, que fornece instruções para a produção da proteína nefrocistina-3. A herança é autossômica recessiva, ou seja, a pessoa precisa herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença. Pais que são portadores de uma única cópia alterada geralmente não apresentam sintomas.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais e sintomas e confirmado por testes genéticos. Os exames disponíveis incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína também pode ser realizada como parte da investigação. Atualmente, há 49 testes genéticos disponíveis e 261 variantes descritas no ClinVar relacionadas a esta condição.[1][2][5]
Tratamento e manejo
O manejo da Síndrome Meckel-like NPHP3-relacionado é multidisciplinar e focado no tratamento dos sintomas e complicações. Não há cura específica. O acompanhamento pode incluir nefrologista (para doença renal), hepatologista (para problemas hepáticos), cardiologista (para alterações cardíacas) e geneticista. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para esta condição, incluindo atendimento em reabilitação para doenças raras. Não há medicamentos específicos aprovados para a síndrome.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas, especialmente o comprometimento renal e hepático. A doença renal crônica estágio 5 pode exigir diálise ou transplante renal. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para melhorar a qualidade de vida e o manejo das complicações.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 9 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 30 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Required for normal ciliary development and function. Inhibits disheveled-1-induced canonical Wnt-signaling activity and may also play a role in the control of non-canonical Wnt signaling which regulates planar cell polarity. Probably acts as a molecular switch between different Wnt signaling pathways. Required for proper convergent extension cell movements
Cell projection, cilium
Nephronophthisis 3
An autosomal recessive disorder resulting in end-stage renal disease. It is characterized by polyuria, polydipsia, anemia. Onset of terminal renal failure occurr significantly later (median age, 19 years) than in juvenile nephronophthisis. Renal pathology is characterized by alterations of tubular basement membranes, tubular atrophy and dilation, sclerosing tubulointerstitial nephropathy, and renal cyst development predominantly at the corticomedullary junction.
Variantes genéticas (ClinVar)
261 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 385 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Meckel-like NPHP3-relacionado
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Expanding the spectrum of CEP55-associated disease to viable phenotypes.
Homozygosity for nonsense variants in CEP55 has been associated with a lethal condition characterized by multinucleated neurons, anhydramnios, renal dysplasia, cerebellar hypoplasia, and hydranencephaly (MARCH syndrome) also known as Meckel-like syndrome. Missense variants in CEP55 have not previously been reported in association with disease. Here we describe seven living individuals from five families with biallelic CEP55 variants. Four unrelated individuals with microcephaly, speech delays, and bilateral toe syndactyly all have a common CEP55 variant c.70G>A p.(Glu24Lys) in trans with nonsense variants. Three siblings are homozygous for a consensus splice site variant near the end of the gene. These affected girls all have severely delayed development, microcephaly, and varying degrees of lissencephaly/pachygyria. Here we compare our seven patients with three previously reported families with a prenatal lethal phenotype (MARCH syndrome/Meckel-like syndrome) due to homozygous CEP55 nonsense variants. Our series suggests that individuals with compound heterozygosity for nonsense and missense variants in CEP55 have a different viable phenotype. We show that homozygosity for a splice variant near the end of the CEP55 gene is also compatible with life.
A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy.
Mutations in genes involved in the cilium-centrosome complex are called ciliopathies. Meckel-Gruber syndrome (MKS) is a ciliopathic lethal autosomal recessive syndrome characterized by genetically and clinically heterogeneous manifestations, including renal cystic dysplasia, occipital encephalocele and polydactyly. Several genes have previously been associated with MKS and MKS-like phenotypes, but there are still genes remaining to be discovered. We have used whole-exome sequencing (WES) to uncover the genetics of a suspected autosomal recessive Meckel syndrome phenotype in a family with 2 affected fetuses. RNA studies and histopathological analysis was performed for further delineation. WES lead to identification of a homozygous nonsense mutation c.256C>T (p.Arg86*) in CEP55 (centrosomal protein of 55 kDa) in the affected fetus. The variant has previously been identified in carriers in low frequencies, and segregated in the family. CEP55 is an important centrosomal protein required for the mid-body formation at cytokinesis. Our results expand the list of centrosomal proteins implicated in human ciliopathies and provide evidence for an essential role of CEP55 during embryogenesis and development of disease.
An ovine hepatorenal fibrocystic model of a Meckel-like syndrome associated with dysmorphic primary cilia and TMEM67 mutations.
Meckel syndrome (MKS) is an inherited autosomal recessive hepatorenal fibrocystic syndrome, caused by mutations in TMEM67, characterized by occipital encephalocoele, renal cysts, hepatic fibrosis, and polydactyly. Here we describe an ovine model of MKS, with kidney and liver abnormalities, without polydactyly or occipital encephalocoele. Homozygous missense p.(Ile681Asn; Ile687Ser) mutations identified in ovine TMEM67 were pathogenic in zebrafish phenotype rescue assays. Meckelin protein was expressed in affected and unaffected kidney epithelial cells by immunoblotting, and in primary cilia of lamb kidney cyst epithelial cells by immunofluorescence. In contrast to primary cilia of relatively consistent length and morphology in unaffected kidney cells, those of affected cyst-lining cells displayed a range of short and extremely long cilia, as well as abnormal morphologies, such as bulbous regions along the axoneme. Putative cilia fragments were also consistently located within the cyst luminal contents. The abnormal ciliary phenotype was further confirmed in cultured interstitial fibroblasts from affected kidneys. These primary cilia dysmorphologies and length control defects were significantly greater in affected cells compared to unaffected controls. In conclusion, we describe abnormalities involving primary cilia length and morphology in the first reported example of a large animal model of MKS, in which we have identified TMEM67 mutations.
Publicações recentes
Whole-body MRI-based long-term evaluation of pediatric NF1 patients without initial tumor burden with evidence of newly developed peripheral nerve sheath tumors.
Prospective cardiovascular magnetic resonance imaging in adults with Alström syndrome: silent progression of diffuse interstitial fibrosis.
Integrating ontologies of rare diseases and radiological diagnosis.
Overcoming the barriers to diagnosis of Morquio A syndrome.
📚 EuropePMCmostrando 3
Expanding the spectrum of CEP55-associated disease to viable phenotypes.
American journal of medical genetics. Part AAn ovine hepatorenal fibrocystic model of a Meckel-like syndrome associated with dysmorphic primary cilia and TMEM67 mutations.
Scientific reportsA nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy.
Clinical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Meckel-like NPHP3-relacionado.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Meckel-like NPHP3-relacionado
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Expanding the spectrum of CEP55-associated disease to viable phenotypes.
- A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy.
- An ovine hepatorenal fibrocystic model of a Meckel-like syndrome associated with dysmorphic primary cilia and TMEM67 mutations.
- Whole-body MRI-based long-term evaluation of pediatric NF1 patients without initial tumor burden with evidence of newly developed peripheral nerve sheath tumors.
- Prospective cardiovascular magnetic resonance imaging in adults with Alström syndrome: silent progression of diffuse interstitial fibrosis.
- Integrating ontologies of rare diseases and radiological diagnosis.
- Overcoming the barriers to diagnosis of Morquio A syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3032(Orphanet)
- OMIM OMIM:267010(OMIM)
- MONDO:0009966(MONDO)
- GARD:4665(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3508640(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
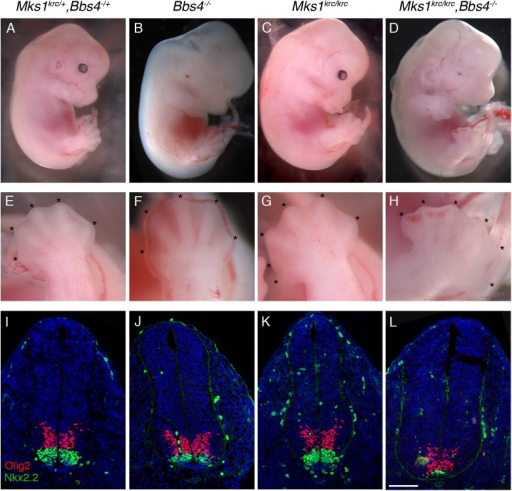
Síndrome Meckel-like NPHP3-relacionado
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata