Síndrome de Perrault que é causada por uma mutação no gene HARS2.
Introdução
O que você precisa saber de cara
Síndrome de Perrault que é causada por uma mutação no gene HARS2.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 2 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 4 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
9 genes identificados com associação a esta condição.
Curadoria gene-doença
fontes oficiaisBifunctional enzyme acting on the peroxisomal fatty acid beta-oxidation pathway. Catalyzes two of the four reactions in fatty acid degradation: hydration of 2-enoyl-CoA (trans-2-enoyl-CoA) to produce (3R)-3-hydroxyacyl-CoA, and dehydrogenation of (3R)-3-hydroxyacyl-CoA to produce 3-ketoacyl-CoA (3-oxoacyl-CoA), which is further metabolized by SCPx. Can use straight-chain and branched-chain fatty acids, as well as bile acid intermediates as substrates
Peroxisome
D-bifunctional protein deficiency
Disorder of peroxisomal fatty acid beta-oxidation.
Catalytic ribonuclease component of mitochondrial ribonuclease P, a complex composed of TRMT10C/MRPP1, HSD17B10/MRPP2 and PRORP/MRPP3, which cleaves tRNA molecules in their 5'-ends (PubMed:18984158, PubMed:25953853, PubMed:34715011). The presence of TRMT10C/MRPP1, HSD17B10/MRPP2 is required to catalyze tRNA molecules in their 5'-ends (PubMed:25953853)
Mitochondrion
Combined oxidative phosphorylation deficiency 54
An autosomal recessive, multisystem disorder with highly variable manifestations resulting from defective mitochondrial transcription and translation. Clinical features include early-onset sensorineural hearing loss, sometimes associated with global developmental delay or primary ovarian failure, peripheral hypertonia, seizures, muscle weakness, behavioral abnormalities, and leukoencephalopathy on brain imaging. Serum lactate may or may not be elevated.
Catalyzes the trans-addition of the three molecules of IPP onto DMAPP to form geranylgeranyl pyrophosphate, an important precursor of carotenoids and geranylated proteins
CytoplasmCytoplasm, perinuclear regionCytoplasm, myofibril, sarcomere, Z line
Muscular dystrophy, congenital hearing loss, and ovarian insufficiency syndrome
An autosomal recessive disorder characterized by early-onset progressive muscle weakness, sensorineural hearing loss, and primary amenorrhea due to ovarian insufficiency. Some patients become wheelchair-bound by the second decade, whereas others have a milder phenotype and maintain independent ambulation into adulthood. Most patients have respiratory insufficiency.
Required for mitochondrial translation, possibly by coordinating the assembly or maintenance of the mitochondrial ribosome (PubMed:23022098, PubMed:25604853)
Mitochondrion
Combined oxidative phosphorylation deficiency 11
A severe, multisystemic, autosomal recessive, disorder characterized by deficiencies of multiple mitochondrial respiratory enzymes leading to neonatal hypotonia and lactic acidosis. Affected individuals may have respiratory insufficiency, foot deformities, or seizures.
Catalyzes the attachment of leucine to its cognate tRNA
Mitochondrion matrix
Perrault syndrome 4
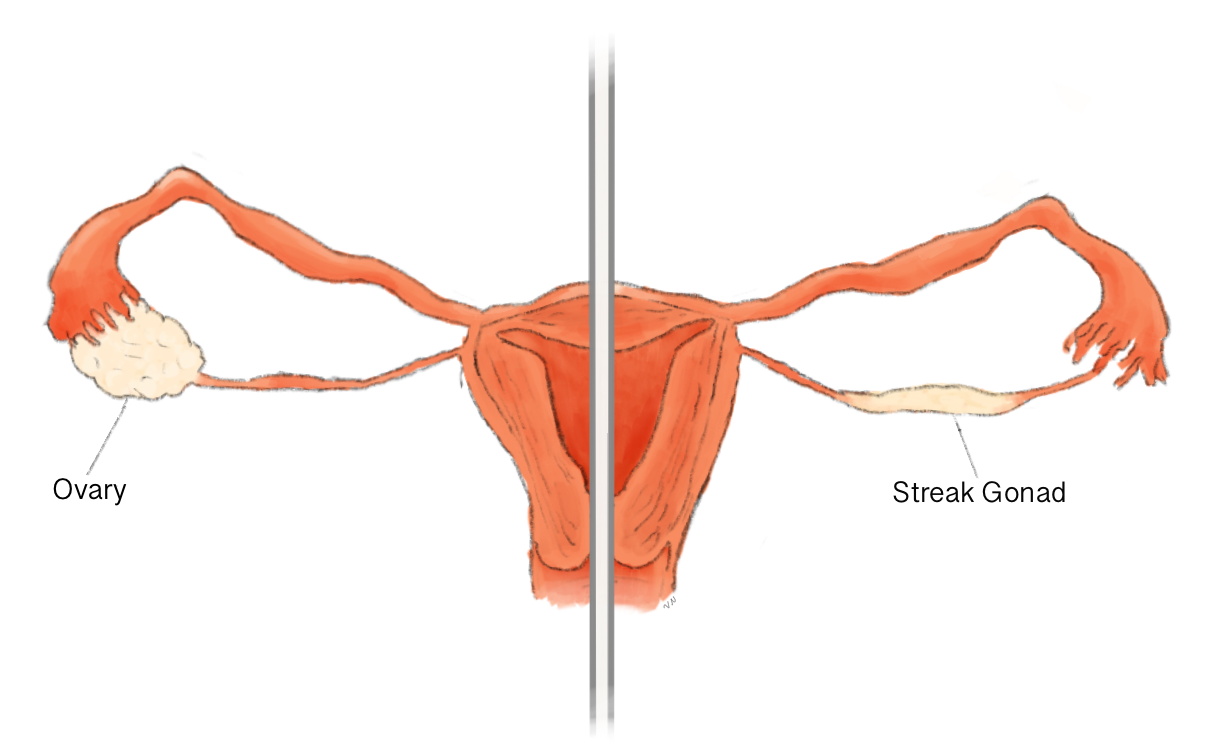
An autosomal recessive, sex-influenced disorder characterized by sensorineural deafness in both males and females, and ovarian dysgenesis in females. Affected females have primary amenorrhea, streak gonads, and infertility, whereas affected males show normal pubertal development and are fertile.
Mitochondrial helicase involved in mtDNA replication and repair (PubMed:12975372, PubMed:15167897, PubMed:17324440, PubMed:18039713, PubMed:18971204, PubMed:25824949, PubMed:26887820, PubMed:27226550). Might have a role in mtDNA repair (PubMed:27226550). Has DNA strand separation activity needed to form a processive replication fork for leading strand synthesis which is catalyzed by the formation of a replisome complex with POLG and mtSDB (PubMed:12975372, PubMed:15167897, PubMed:18039713, PubMe
Mitochondrion matrix, mitochondrion nucleoidMitochondrion inner membrane
Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant, 3
A disorder characterized by progressive weakness of ocular muscles and levator muscle of the upper eyelid. In a minority of cases, it is associated with skeletal myopathy, which predominantly involves axial or proximal muscles and which causes abnormal fatigability and even permanent muscle weakness. Ragged-red fibers and atrophy are found on muscle biopsy. A large proportion of chronic ophthalmoplegias are associated with other symptoms, leading to a multisystemic pattern of this disease. Additional symptoms are variable, and may include cataracts, hearing loss, sensory axonal neuropathy, ataxia, depression, hypogonadism, and parkinsonism.
Protease component of the ClpXP complex that cleaves peptides and various proteins in an ATP-dependent process. Has low peptidase activity in the absence of CLPX. The ClpXP complex can degrade CSN1S1, CSN2 and CSN3, as well as synthetic peptides (in vitro) and may be responsible for a fairly general and central housekeeping function rather than for the degradation of specific substrates (PubMed:11923310, PubMed:15522782). Cleaves PINK1 in the mitochondrion (PubMed:22354088)
Mitochondrion matrix
Perrault syndrome 3
An autosomal recessive, sex-influenced disorder characterized by sensorineural deafness in both males and females, and ovarian dysgenesis in females. Affected females have primary amenorrhea, streak gonads, and infertility, whereas affected males show normal pubertal development and are fertile. A spectrum of additional clinical features, including cerebellar ataxia, learning disability, and peripheral neuropathy, have been described in some PRLTS3 affected individuals.
Possible ATPase (PubMed:15653697) involved in DNA replication, may facilitate loading of CDC45 onto pre-replication complexes (PubMed:20065034) An aminoacyl-tRNA editing enzyme that deacylates mischarged D-aminoacyl-tRNAs. Also deacylates mischarged glycyl-tRNA(Ala), protecting cells against glycine mischarging by AlaRS. Acts via tRNA-based rather than protein-based catalysis; rejects L-amino acids rather than detecting D-amino acids in the active site. By recycling D-aminoacyl-tRNA to D-amino a
NucleusCytoplasm
Probable GTPase that plays a role in the mitochondrial ribosomal small subunit assembly. Specifically binds the 12S mitochondrial rRNA (12S mt-rRNA) to a 33 nucleotide section delineating the 3' terminal stem-loop region. May act as a chaperone that protects the 12S mt-rRNA on the 28S mitoribosomal subunit during ribosomal small subunit assembly
Mitochondrion matrixMitochondrion inner membrane
Perrault syndrome 6
A form of Perrault syndrome, a sex-influenced disorder characterized by sensorineural deafness in both males and females, and ovarian dysgenesis in females. Affected females have primary amenorrhea, streak gonads, and infertility, whereas affected males show normal pubertal development and are fertile. PRLTS6 inheritance is autosomal recessive.
Variantes genéticas (ClinVar)
177 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.214 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
21 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Perrault tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Mostrando amostra de 14 publicações de um total de 98
A novel CLPP variant in a Pakistani family with Perrault syndrome associated with recurrent fevers.
Perrault syndrome (PRLTS) is an autosomal recessive disease with sensorineural hearing loss and ovarian dysfunction in girls, and either a fluctuating neurological phenotype or not. PRLTS type 2 is known to be caused by pathogenic variants of the CLPP gene that encodes mitochondrial ATP-dependent protease. This paper involved clinical and genetic studies on a Pakistani family with PRLTS. Whole-exome sequencing identified a novel homozygous CLPP missense mutation (NM_006012.4: c.250 A > C; p.Ile84Leu). Its pathogenicity was assessed with the help of multiple sequence alignment, AlphaFold protein modeling, and docking with CLPX with the help of ClusPro. Auditory brainstem responses and tympanometry were in clinical assessment. The individuals were found to have a uniform phenotype of severe sensorineural hearing loss, mild intellectual disability, ataxia and frequent fever. There was one patient in whom the unilateral Eustachian tube dysfunction was hinted at by Tympanometry. At the molecular level, the identified CLPP variant involved a highly conserved residue. Structural modeling showed preserved protein architecture, whereas docking simulations revealed disrupted CLPP-CLPX interaction, suggesting a basis for impaired proteostasis. We report a novel CLPP missense variant (p.I84L) in a Pakistani family with PRLTS, expanding the mutational spectrum of CLPP. To the best of our knowledge, recurrent fever was reported in PRLTS for the first time, which expanded the PRLTS phenotype spectrum.
CLPP Gene Variants Causing Perrault Syndrome Type 3 in Han Chinese Families: A Genotype-Phenotype Study.
Perrault syndrome is a rare autosomal recessive disorder characterized by sensorineural hearing loss (SNHL) and primary ovarian insufficiency (POI) secondary to ovarian dysgenesis. However, the mutation spectrum of disease-causing genes for Perrault syndrome in the Chinese population remains poorly understood. In this study, we report on two Chinese families with Perrault syndrome type 3 caused by novel CLPP gene variants. We also conducted a comprehensive literature review of CLPP gene variants in Perrault syndrome type 3 to elucidate genotype-phenotype associations. Using Whole Genome Sequencing (WGS) data, two pedigrees with Perrault syndrome type 3 were ascertained in the Chinese Deafness Genetics Cohort through genotype-driven analysis. Variants were validated using Sanger sequencing and copy number quantification methods. In vitro analysis of splice site variants in the CLPP gene using the minigene assay. Two Han Chinese families were ascertained: one with compound heterozygous variants (c.270 + 1G > C and c.355A > C [p. Ile119Leu]) and the other with missense variant (c.400G > C [p. Asp134His]) together with a large deletion in CLPP. In vitro minigene assays confirmed that the c.270 + 1G > C variant causes intron 2 retention and an alternative 5' splice site in exon 2, leading to protein alteration. Among 33 Perrault syndrome type 3 patients in literature, 97% (31/32) had hearing loss, 55% (16/29) neurological disease, and 71% (15/21) females had POI. Including our 4 novel variants, 21 pathogenic CLPP gene variants have been reported, with 57% (12/21) missense and 43% (9/21) truncating variants, mainly in the ATP-dependent Clp protease proteolytic subunit. Biallelic truncating or missense plus truncating genotypes showed higher rates of neurological disease (p = 0.001), but no significant difference in hearing loss incidence compared to biallelic missense genotypes was observed. This study highlights the challenges in diagnosing Perrault syndrome due to its genetically and clinically heterogeneity. By exploring novel variants and establishing genotype-phenotype correlations, we aim to improve the genetic diagnosis and consultation for this complex disorder. The purpose of this overview is to: 1.. Briefly describe the clinical characteristics of Perrault syndrome; 2.. Review the genetic causes of Perrault syndrome; 3.. Review the differential diagnosis of Perrault syndrome with a focus on genetic conditions; 4.. Provide an evaluation strategy to identify the genetic cause of Perrault syndrome in a proband (when possible); 5.. Review management of Perrault syndrome; 6.. Inform genetic counseling of family members of an individual with Perrault syndrome.
Perrault syndrome: The Way Forward After Genetic Counselling?
A female, term neonate, born via vaginal delivery to a G5P1D1A3 hypothyroid mother with a history of an elder sibling being homozygous for HSD17B4 mutation, diagnosed while working up his progressive neurological disorder and succumbing to the same. The family screening revealed that both parents were heterozygous carriers of the same mutation in the gene HSD17B4 After genetic counselling, amniocentesis revealed the fetus to be having homozygosity for the same mutation. In view of precious pregnancy, normal antenatal scans and investigations, the pregnancy was continued, and baby was born with a birth weight of 2.65 kg and had a smooth perinatal transition. Parents were counselled regarding the course of the illness, possible complications and the need for regular follow-up. Ultrasound of the abdomen, pelvis and head was normal in the neonatal period. She was vaccinated as per the national schedule and gaining weight normally.
Exome sequencing reveals pathogenic mutations in the LARS2 and HSD17B4 genes associated with Perrault syndrome and D-bifunctional protein deficiency in Moroccan families.
Syndromic hearing loss (SHL) is characterized by hearing impairment accompanied by other clinical manifestations, reaching over 400 syndromes. Early and accurate diagnosis is essential to understand the progression of hearing loss and associated systemic complications. In this study, we investigated the genetic etiology of sensorineural hearing loss in three Moroccan patients using whole exome sequencing (WES). The results revealed in two families Perrault syndrome caused by LARS2, p. Asn153His; p. Thr629Met compound heterozygous variants in two siblings in one family; and p. Thr522Asn, a homozygous variant in two sisters in another. The patient in the third family was diagnosed with D-bifunctional protein deficiency (D-BPD), linked to compound heterozygous mutations p. Asn457Tyr and p. Val643Argfs*5 in HSD17B4. Molecular dynamic simulation results showed that Val643Argfs*5 does not prevent HSD17B4 protein from binding to the PEX5 receptor, but further studies are recommended to verify its effect on HSD17B4 protein functionality. These results highlight the effectiveness of WES in identifying pathogenic mutations involved in heterogeneous disorders and the usefulness of bioinformatics in predicting their effects on protein structure.
[Analysis of perrault syndrome caused by pathogenic variants in LARS2 and HARS2 genes].
Objective: To investigate the molecular etiology of Perrault syndrome by analyzing the clinical phenotype and pathogenic gene variants of 2 male patients with bilateral severe sensorineural deafness. Methods: Two male patients with Perrault syndrome characterized by severe sensonrineual deafness adimitted to the First Affiliated Hospital of Zhengzhou University between February 2021 and March 2022 were selected, and the clinical phenotype and pathogenic gene variants of them and their family members were summarized. The whole exome sequencing technology was used to screen the pathogenic variants of the probands, and the candidate variants were determined by combining with clinical phenotype. The probands and their family members were verified by the Sanger sequencing method. Results: The whole exome sequencing results showed that the proband of family 1 had a compound heterozygous variants of the LARS2 (NM_015340.4) gene c.1565C>A (p.Thr522Asn) and c.1079T>C (p.Ile360Thr). The reported pathogenic variant c.1565C>A came from the mother, and the novel variant c.1079T>C came from the father. The second proband harbored compound heterozygous variants of HARS2 gene (NM_012208.4) c.1273C>T (p.Arg425Trp) and c.1403G>C (p.Gly468Ala), with the former from the proband's mother, the latter from the father. The c.1273C>T was novel and c.1403G>C was the reported pathogenic variant. All above variants were respectively classified as pathogenic, uncertain significance, uncertain significance and likely pathogenic based on the ACMG guidelines. Conclusion: This study expands the mutational spectrum of LARS2 and HARS2 genes, which highlights that genetic testing plays an important role in the early diagnosis of syndromic deafness. 目的: 分析并揭示Perrault综合征患者的分子病因。 方法: 选取郑州大学第一附属医院2021年2月至2022年3月收治的2例以双耳重度感音神经性听力损失为特征的男性Perrault综合征患者,总结患者及其家系的临床表型及基因变异,应用全外显子测序(whole exome sequencing,WES)技术对先证者进行致病变异筛选,结合临床表型确定候选基因及致病位点,用Sanger测序法对先证者及其家系成员进行验证。 结果: WES结果显示家系1的先证者存在LARS2基因(NM_015340.4)c.1565C>A(p.Thr522Asn)和c.1079T>C(p.Ile360Thr)的复合杂合变异,c.1565C>A来自先证者母亲,c.1079T>C来自先证者父亲。其中,c.1565C>A为已报道的致病变异,c.1079T>C为新变异。家系2先证者携带HARS2基因(NM_012208.4)c.1273C>T(p.Arg425Trp)和c.1403G>C(p.Gly468Ala)的复合杂合变异,c.1273C>T来自母亲,c.1403G>C来自父亲,c.1273C>T为新变异,c.1403G>C为已报道可能致病变异。参考美国医学遗传学与基因组学学会基因变异分类标准,上述变异分别评级为致病、意义未明、意义未明和可能致病。 结论: 本研究扩大了LARS2和HARS2基因的突变谱,基因检测在综合征型耳聋的早期诊断中发挥着重要作用。.
Publicações recentes
Perrault Syndrome Presenting With Progressive Ataxia and the Hot Cross Bun Sign.
Perrault syndrome unmasked: genomic reclassification of a Fabry-like CKDx phenotype.
Novel LARS2 variants in patients with Perrault syndrome: expanding the genetic spectrum and phenotypic heterogeneity.
Expanding the genotypic spectrum of combined oxidative phosphorylation deficiency 54.
Patient-derived TWNK variants recapitulate multisystem Perrault syndrome pathology in a mouse model.
📚 EuropePMC84 artigos no totalmostrando 14
A novel CLPP variant in a Pakistani family with Perrault syndrome associated with recurrent fevers.
Clinica chimica acta; international journal of clinical chemistryCLPP Gene Variants Causing Perrault Syndrome Type 3 in Han Chinese Families: A Genotype-Phenotype Study.
Human genomicsExome sequencing reveals pathogenic mutations in the LARS2 and HSD17B4 genes associated with Perrault syndrome and D-bifunctional protein deficiency in Moroccan families.
Molecular biology reportsPerrault syndrome: The Way Forward After Genetic Counselling?
BMJ case reports[Analysis of perrault syndrome caused by pathogenic variants in LARS2 and HARS2 genes].
Zhonghua er bi yan hou tou jing wai ke za zhi = Chinese journal of otorhinolaryngology head and neck surgery[Genetic analysis of a child with D bifunctional protein deficiency born to a consanguineous pedigree].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsA known pathogenic variant in the essential mitochondrial translation gene RMND1 causes a Perrault-like syndrome with renal defects.
Clinical geneticsBiallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms.
American journal of medical genetics. Part AA homozygous missense variant in HSD17B4 identified in a consanguineous Chinese Han family with type II Perrault syndrome.
BMC medical geneticsA homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome.
Human molecular geneticsAn Application of NGS for Molecular Investigations in Perrault Syndrome: Study of 14 Families and Review of the Literature.
Human mutationMutations of SGO2 and CLDN14 collectively cause coincidental Perrault syndrome.
Clinical geneticsExpanding the genotypic spectrum of Perrault syndrome.
Clinical geneticsFirst independent replication of the involvement of LARS2 in Perrault syndrome by whole-exome sequencing of an Italian family.
Journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Perrault tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Perrault tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A novel CLPP variant in a Pakistani family with Perrault syndrome associated with recurrent fevers.
- CLPP Gene Variants Causing Perrault Syndrome Type 3 in Han Chinese Families: A Genotype-Phenotype Study.
- Perrault syndrome: The Way Forward After Genetic Counselling?
- Exome sequencing reveals pathogenic mutations in the LARS2 and HSD17B4 genes associated with Perrault syndrome and D-bifunctional protein deficiency in Moroccan families.
- [Analysis of perrault syndrome caused by pathogenic variants in LARS2 and HARS2 genes].Zhonghua er bi yan hou tou jing wai ke za zhi = Chinese journal of otorhinolaryngology head and neck surgery· 2023· PMID 38186093mais citado
- Perrault Syndrome Presenting With Progressive Ataxia and the Hot Cross Bun Sign.
- Perrault syndrome unmasked: genomic reclassification of a Fabry-like CKDx phenotype.
- Novel LARS2 variants in patients with Perrault syndrome: expanding the genetic spectrum and phenotypic heterogeneity.
- Expanding the genotypic spectrum of combined oxidative phosphorylation deficiency 54.
- Patient-derived TWNK variants recapitulate multisystem Perrault syndrome pathology in a mouse model.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:642976(Orphanet)
- OMIM OMIM:614926(OMIM)
- MONDO:0013972(MONDO)
- GARD:15882(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Perrault tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)