A Síndrome de Surdez e Infertilidade (SSI) é uma síndrome muito rara que combina surdez sensorioneural com infertilidade masculina.
Introdução
O que você precisa saber de cara
A Síndrome de Surdez e Infertilidade (SSI) é uma síndrome muito rara que combina surdez sensorioneural com infertilidade masculina.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
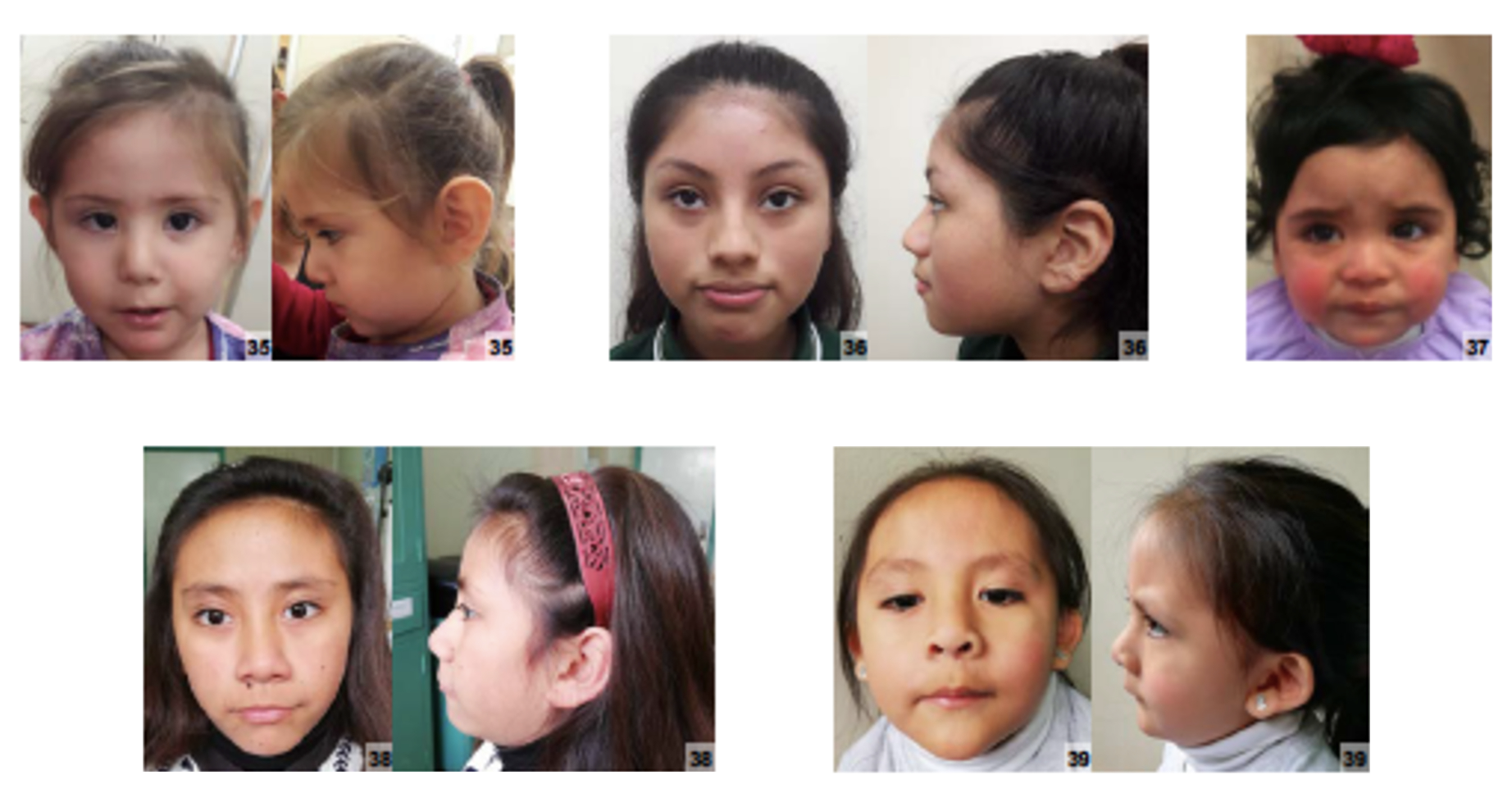
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 11 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Pore-forming subunit of the CatSper complex, a sperm-specific voltage-gated calcium channel, that plays a central role in calcium-dependent physiological responses essential for successful fertilization, such as sperm hyperactivation, acrosome reaction and chemotaxis towards the oocyte
Cell projection, cilium, flagellum membrane
Deafness-infertility syndrome
Characterized by deafness and infertility and is caused by large contiguous gene deletions at 15q15.3 that removes both STRC and CATSPER2 genes.
Essential to the formation of horizontal top connectors between outer hair cell stereocilia
Cell surfaceCell projection, kinociliumCell projection, stereocilium
Deafness, autosomal recessive, 16
A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information.
Variantes genéticas (ClinVar)
178 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 16 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome surdez-infertilidade
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Natural History of Sensorineural Hearing Loss in Children With STRC Mutations.
The most common genes responsible for autosomal recessive nonsyndromic hearing loss (AR-NSHL) are GJB2 and STRC. STRC mutations are associated with mild-to-moderate sensorineural (SNHL) hearing loss and a lack of progression. However, our institutional experience suggested otherwise, prompting this review. A 10-year retrospective chart review was performed at a tertiary children's hospital after the University of Iowa added STRC to its OtoSCOPER panel in 2013. Subjects with positive OtoSCOPER results underwent audiologic review. Hearing progression was defined based on pure-tone average changes, and mutation subtypes were categorized. Of 354 subjects undergoing OtoSCOPER testing, 181 (51.1%) carried a pathogenic mutation; GJB2 (28.7%) and STRC (16.6%) were most common. The STRC cohort included 30 subjects (21 males, 9 females) with hearing loss severity classifiable in 26 subjects and the highest proportion in the mild-to-moderate range (n = 46 ears; 88.5%). Hearing progression was observed in 12/24 subjects (20 ears: 8 bilateral, 4 unilateral). Median annual progression was 1.1 dB (range -3.5 to 18.7 dB). Two STRC subjects had substantial progression requiring cochlear implantation (one performed, one recommended). Genetic subtyping revealed seven categories, including six males with STRC/CATSPER2 deletions (deafness-infertility syndrome). No association between subtype and severity or progression was identified. STRC is the second most common cause of childhood NSHL and the leading contributor to mild-to-moderate SNHL. Unlike most published literature, 50% of our STRC cohort exhibited progression, and 17.6% of progressing subjects had substantial unilateral loss. We recommend long-term audiometric monitoring and standardized genomic reporting for this population.
Determination of carriers of deafness-infertility syndrome in Peru.
The prevalence of deafness-infertility syndrome (DIS) is approximately 1%. Genetic heterogeneity is one cause of homozygous copy number variants (CNVs) involving the CATSPER2 and STRC genes, which are associated with DIS and male infertility. Because the prevalence of DIS in Peru is unknown, we aimed to determine the frequency of carriers of DIS-related genes. In this descriptive crossover study, we evaluated the clinical histories and chromosomal microarray analysis results of patients at the Instituto Nacional de Salud del Niño Breña from 2015 to 2022. All patients with CNVs involving the CATSPER2 and STRC genes were included, and the frequencies of carriers and affected patients were determined using Hardy‒Weinberg equilibrium. Relative frequency differences were calculated using the chi-square test with goodness-of-fit for natural regions and poverty groups in Peru. Of 2,142 patients screened, 35 met the inclusion criteria; according to the results, approximately 367,364 people were estimated to be DIS carriers in Peru, and approximately 57,442 people had deafness and infertility. The proportion of carriers in Peru was similar to that observed in other population studies. Additionally, people in regions with higher poverty rates exhibited a greater carrier frequency, suggesting that a patient's region of origin could be a criterion for DIS screening.
NGS sequencing reveals the cause of hearing loss in a group of Polish patients with an isolated, non-DFNB1 hearing loss.
The etiology of hearing loss (HL) is heterogeneous. It is estimated that 50-60% of the cases have a genetic background, with the other part being environmental. Isolated HL is responsible for nearly two-thirds of congenital cases, and the remaining part accounts for syndromic forms (SHL). The study aim was to examine the molecular basis of HL in 48 Polish patients with isolated, non-DFNB1 hearing loss using the targeted next-generation sequencing technique (NGS). The molecular cause of the HL was defined in 39.6% (19/48) of patients. In thirteen genes, we identified causative variants, including six novel ones: p.Gly1326Val (STRC), p.Pro104ThrfsTer2 (MYO6), p.Tyr186Ter (GATA3), p.Ile1584SerfsTer12 (MYO15A), p.Pro559Leu, and p.Glu542del (CDH23). The pathogenic status of novel variants was assessed by using bioinformatic tools and the ACMG recommendations. The most frequent genetic variants were the STRC gene deletions and point variants in Usher syndrome genes. For 36.8% of patients, the molecular diagnosis suggested SHL (Deafness-Infertility Syndrome (DIS), Hypoparathyroidism, Sensorineural Deafness and Renal Disease (HDR), Usher, Perrault and Waardenburg syndromes). The obtained results confirmed the heterogeneity of the molecular basis of HL in Polish patients and the usefulness of the NGS technique as a diagnostic tool.
Unraveling the Genetic Basis of Combined Deafness and Male Infertility Phenotypes through High-Throughput Sequencing in a Unique Cohort from South India.
The co-occurrence of sensorineural hearing loss and male infertility has been reported in several instances, suggesting potential shared genetic underpinnings. One such example is the contiguous gene deletion of CATSPER2 and STRC genes, previously associated with deafness-infertility syndrome (DIS) in males. Fifteen males with both hearing loss and infertility from southern India after exclusion for the DIS contiguous gene deletion and the FOXI1 gene mutations are subjected to exome sequencing. This resolves the genetic etiology in four probands for both the phenotypes; In the remaining 11 probands, two each conclusively accounted for deafness and male infertility etiologies. Genetic heterogeneity is well reflected in both phenotypes. Four recessive (TRIOBP, SLC26A4, GJB2, COL4A3) and one dominant (SOX10) for the deafness; six recessive genes (LRGUK, DNAH9, ARMC4, DNAH2, RSPH6A, and ACE) for male infertility can be conclusively ascribed. LRGUK and RSPH6A genes are implicated earlier only in mice models, while the ARMC4 gene is implicated in chronic destructive airway diseases due to primary ciliary dyskinesia. This study would be the first to document the role of these genes in the male infertility phenotype in humans. The result suggests that deafness and infertility are independent events and do not segregate together among the probands.
Leveraging Unique Chromosomal Microarray Probes to Accurately Detect Copy Number at the Highly Homologous 15q15.3 Deafness-Infertility Syndrome Locus.
Biallelic deletions at 15q15.3, including STRC and CATSPER2, cause autosomal recessive deafness-infertility syndrome (DIS), while biallelic deletions of STRC alone cause nonsyndromic hearing loss. These deletions are among the leading genetic causes of mild-moderate hearing loss, but their detection using chromosomal microarray (CMA) is impeded by a tandem duplication containing highly homologous pseudogenes. We sought to assess copy number variant (CNV) detection in this region by a commonly-employed CMA platform. Twenty-two specimens with known 15q15.3 CNVs, determined by droplet digital PCR (ddPCR), were analyzed by CMA. To investigate the impact of pseudogene homology on CMA performance, a probe-level analysis of homology was performed, and log2 ratios of unique and pseudogene-homologous probes compared. Assessment of 15q15.3 CNVs by CMA compared to ddPCR revealed 40.9% concordance, with frequent mis-assignment of zygosity by the CMA automated calling software. Probe-level analysis of pseudogene homology suggested that probes with high homology contributed to this discordance, with significant differences in log2 ratios between unique and pseudogene-homologous CMA probes. Two clusters containing several unique probes could reliably detect CNVs involving STRC and CATSPER2, despite the noise of surrounding probes, discriminating between homozygous vs heterozygous losses and complex rearrangements. CNV detection by these probe clusters showed 100% concordance with ddPCR. Manual analysis of clusters containing unique CMA probes without significant pseudogene homology improves CNV detection and zygosity assignment in the highly homologous DIS region. Incorporation of this method into CMA analysis and reporting processes can improve DIS diagnosis and carrier detection.
Publicações recentes
Natural History of Sensorineural Hearing Loss in Children With STRC Mutations.
Determination of carriers of deafness-infertility syndrome in Peru.
NGS sequencing reveals the cause of hearing loss in a group of Polish patients with an isolated, non-DFNB1 hearing loss.
Unraveling the Genetic Basis of Combined Deafness and Male Infertility Phenotypes through High-Throughput Sequencing in a Unique Cohort from South India.
Maximizing the Detection of Copy Number Variants in the Highly Homologous Deafness-Infertility Syndrome Locus in Standard-of-care Testing.
📚 EuropePMC5 artigos no totalmostrando 14
Natural History of Sensorineural Hearing Loss in Children With STRC Mutations.
The LaryngoscopeDetermination of carriers of deafness-infertility syndrome in Peru.
Orphanet journal of rare diseasesNGS sequencing reveals the cause of hearing loss in a group of Polish patients with an isolated, non-DFNB1 hearing loss.
Journal of applied geneticsUnraveling the Genetic Basis of Combined Deafness and Male Infertility Phenotypes through High-Throughput Sequencing in a Unique Cohort from South India.
Advanced genetics (Hoboken, N.J.)Maximizing the Detection of Copy Number Variants in the Highly Homologous Deafness-Infertility Syndrome Locus in Standard-of-care Testing.
Clinical chemistryLeveraging Unique Chromosomal Microarray Probes to Accurately Detect Copy Number at the Highly Homologous 15q15.3 Deafness-Infertility Syndrome Locus.
Clinical chemistryCharacterization of the promoter region of the murine Catsper2 gene.
FEBS open bioFrequency of the STRC-CATSPER2 deletion in STRC-associated hearing loss patients.
Scientific reportsPrevalence and Characteristics of STRC Gene Mutations (DFNB16): A Systematic Review and Meta-Analysis.
Frontiers in geneticsAudiologic Phenotype and Progression in Pediatric STRC-Related Autosomal Recessive Hearing Loss.
The LaryngoscopeSignificant Mendelian genetic contribution to pediatric mild-to-moderate hearing loss and its comprehensive diagnostic approach.
Genetics in medicine : official journal of the American College of Medical GeneticsPrenatal cytogenomic identification and molecular refinement of compound heterozygous STRC deletion breakpoints.
Molecular genetics & genomic medicineMaternal uniparental disomy of chromosome 15 and concomitant STRC and CATSPER2 deletion-mediated deafness-infertility syndrome.
American journal of medical genetics. Part ADFNB16 is a frequent cause of congenital hearing impairment: implementation of STRC mutation analysis in routine diagnostics.
Clinical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome surdez-infertilidade.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome surdez-infertilidade
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Natural History of Sensorineural Hearing Loss in Children With STRC Mutations.
- Determination of carriers of deafness-infertility syndrome in Peru.
- NGS sequencing reveals the cause of hearing loss in a group of Polish patients with an isolated, non-DFNB1 hearing loss.
- Unraveling the Genetic Basis of Combined Deafness and Male Infertility Phenotypes through High-Throughput Sequencing in a Unique Cohort from South India.
- Leveraging Unique Chromosomal Microarray Probes to Accurately Detect Copy Number at the Highly Homologous 15q15.3 Deafness-Infertility Syndrome Locus.
- Maximizing the Detection of Copy Number Variants in the Highly Homologous Deafness-Infertility Syndrome Locus in Standard-of-care Testing.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:94064(Orphanet)
- OMIM OMIM:611102(OMIM)
- MONDO:0012621(MONDO)
- GARD:11911(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55783802(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome surdez-infertilidade
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub