A síndrome de Weill-Marchesani (SWM) é uma doença rara caracterizada por baixa estatura, dedos curtos, rigidez nas articulações e problemas característicos nos olhos, incluindo lentes oculares pequenas e arredondadas, lentes fora do lugar, grau alto de miopia e glaucoma.
Introdução
O que você precisa saber de cara
A síndrome de Weill-Marchesani (SWM) é uma doença rara caracterizada por baixa estatura, dedos curtos, rigidez nas articulações e problemas característicos nos olhos, incluindo lentes oculares pequenas e arredondadas, lentes fora do lugar, grau alto de miopia e glaucoma.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 23 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 61 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Metalloprotease that participate in microfibrils assembly. Microfibrils are extracellular matrix components occurring independently or along with elastin in the formation of elastic tissues
Secreted, extracellular space, extracellular matrix
Weill-Marchesani syndrome 1
A rare connective tissue disorder characterized by short stature, brachydactyly, joint stiffness, and eye abnormalities including microspherophakia, ectopia lentis, severe myopia and glaucoma.
Ceramide synthase that catalyzes the transfer of the acyl chain from acyl-CoA to a sphingoid base, with high selectivity toward very- and ultra-long-chain fatty acyl-CoA (chain length greater than C22) (PubMed:17977534, PubMed:22038835, PubMed:26887952). N-acylates sphinganine and sphingosine bases to form dihydroceramides and ceramides in de novo synthesis and salvage pathways, respectively (PubMed:17977534, PubMed:22038835, PubMed:26887952). It is crucial for the synthesis of ultra-long-chain
Endoplasmic reticulum membrane
Ichthyosis, congenital, autosomal recessive 9
A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs.
Structural component of the 10-12 nm diameter microfibrils of the extracellular matrix, which conveys both structural and regulatory properties to load-bearing connective tissues (PubMed:15062093, PubMed:1860873). Fibrillin-1-containing microfibrils provide long-term force bearing structural support (PubMed:27026396). In tissues such as the lung, blood vessels and skin, microfibrils form the periphery of the elastic fiber, acting as a scaffold for the deposition of elastin (PubMed:27026396). In
SecretedSecreted, extracellular space, extracellular matrix
Marfan syndrome
A hereditary disorder of connective tissue that affects the skeletal, ocular, and cardiovascular systems. A wide variety of skeletal abnormalities occurs with Marfan syndrome, including scoliosis, chest wall deformity, tall stature, abnormal joint mobility. Ectopia lentis occurs in most of the patients and is almost always bilateral. The leading cause of premature death is progressive dilation of the aortic root and ascending aorta, causing aortic incompetence and dissection. Neonatal Marfan syndrome is the most severe form resulting in death from cardiorespiratory failure in the first few years of life.
May play an integral structural role in elastic-fiber architectural organization and/or assembly
Secreted, extracellular space, extracellular matrix
Glaucoma 3, primary congenital, D
An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor.
Secreted, extracellular space, extracellular matrix
Weill-Marchesani syndrome 4
An autosomal recessive syndrome characterized by lenticular myopia, ectopia lentis, glaucoma, spherophakia and short stature. Brachydactyly and decreased joint flexibility are present in some patients.
Variantes genéticas (ClinVar)
4.919 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 897 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
10 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Weill-Marchesani
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A novel homozygous ADAMTS10 frameshift variant in Weill-Marchesani syndrome in a Chinese family.
Weill–Marchesani syndrome (WMS) is a rare connective tissue disorder. The main clinical features include short stature with a stocky build, brachydactyly, joint stiffness, and microspherophakia. The syndrome can be inherited in an autosomal dominant manner due to heterozygous pathogenic variants in FBN1, as well as in an autosomal recessive manner associated with biallelic pathogenic variants in ADAMTS10, ADAMTS17, or LTBP2. Despite differences in inheritance patterns, the clinical manifestations are consistent. Therefore, clinical diagnosis requires genetic testing of the proband to determine the genotype, confirm the diagnosis. This study collected blood samples from a child with Weill–Marchesani syndrome (WMS) and their family members from a Chinese family. Comprehensive ophthalmologic and systemic examinations were performed. Whole-exome sequencing (WES) was conducted to detect genetic variants, and bioinformatics tools were used to screen for potential pathogenic variants. Suspected variants were validated by Sanger sequencing and comprehensively evaluated in combination with clinical data. Using the keywords “ADAMTS10” and “Weill–Marchesani syndrome,” relevant cases were retrieved from databases such as PubMed, CNKI (China National Knowledge Infrastructure), and Wanfang Medical. The genotypes and clinical manifestations of reported cases were analyzed and summarized. The proband presented with clinical features including short stature, brachydactyly, and ocular abnormalities. Ocular manifestations included high myopia, microspherophakia, and glaucoma. Whole-exome sequencing revealed a homozygous frameshift duplication variant in the ADAMTS10: NM_030957.4:c.1560_1575dup; p.Ile526Valfs*51. Both phenotypically normal parents were heterozygous carriers of the same variant. According to the variant interpretation principles of the Human Gene Mutation Database (HGMD) and the guidelines of the American College of Medical Genetics and Genomics (ACMG), this variant was classified as pathogenic. A review of the literature indicated that WMS associated with ADAMTS10 variants exhibits complex and heterogeneous clinical phenotypes. A novel homozygous frameshift variant of ADAMTS10is identified in a WMS family with a history of consanguineous marriage. Our study expands the range of variations associated with WMS and deepens our understanding of pathology in disease associated with ADAMTS10 variants. The online version contains supplementary material available at 10.1186/s12920-026-02308-7.
A novel compound heterozygous mutation in ADAMTS17 identified in a Chinese family with Weill-Marchesani syndrome.
To investigate the genetic basis of Weill-Marchesani syndrome (WMS) in a Chinese family and clarify the pathogenic mechanism of novel ADAMTS17 mutations. Comprehensive clinical assessments and genetic analyses were performed on a Chinese family with two affected siblings. Whole-exome sequencing (WES) was conducted for the proband and other family members. Bioinformatics tools were used to evaluate the conservation, predicted pathogenicity, and structural effects of the identified ADAMTS17 variants. In addition, protein structure modeling was applied to assess the functional impacts of the mutations. The proband (a 32-year-old male) and his elder sister (42y) presented typical clinical features of WMS, including short stature, brachydactyly, high myopia, ectopia lentis, and secondary glaucoma. WES identified a novel compound heterozygous mutation in ADAMTS17: a splicing mutation (c.451-2A>G) inherited from the father and a missense mutation (c.1043G>A; p.C348Y) inherited from the mother. The splicing mutation disrupted normal mRNA splicing and processing, leading to premature translation termination. The missense mutation, which is located in the metalloprotease catalytic domain, was predicted to abolish a critical disulfide bond, thereby impairing protein stability. Both mutations exhibited high evolutionary conservation and were predicted to be pathogenic by multiple bioinformatics algorithms. A novel compound heterozygous mutation in ADAMTS17 is identified in this WMS-affected Chinese family, and its pathogenicity is verified via bioinformatics analysis and protein structural modeling. These findings are expected to facilitate the genetic diagnosis of WMS and deepen the understanding of its molecular pathogenesis.
A unique case of microspherophakia in adult Refsum disease.
Adult Refsum disease (RD) is a rare autosomal recessive peroxisomal disorder with an estimated prevalence of fewer than 1 in 1 million. The ocular manifestations result from the accumulation of phytanic acid, leading to retinitis pigmentosa, attenuated retinal vessels, bone spicule pigmentation, optic disc pallor, and macular involvement in advanced stages. To date, there has been no documented association between RD and microspherophakia, a rare lens abnormality more commonly linked to Weill-Marchesani syndrome. Microspherophakia is characterized by a small lens diameter, increased anteroposterior thickness, and a visible lens equator upon full mydriasis. We present a unique case of concurrent RD and microspherophakia.
Combined ADAMTS10 and ADAMTS17 inactivation exacerbates bone shortening and skin phenotypes.
Weill-Marchesani syndrome (WMS) is characterized by severe short stature, joint contractures, tight skin, heart valve and eye anomalies. WMS is caused by biallelic mutations in ADAMTS10, ADAMTS17, or LTBP2, or mono-allelic mutations in FBN1 Because bone growth is driven by chondrocyte proliferation and hypertrophy in the growth plates, the genetics of WMS suggests that the affected extracellular matrix (ECM) proteins contribute to chondrocyte and growth plate function. Here, we show that Adamts10;Adamts17 double knockout (DKO) mice have significant postnatal lethality and exacerbated bone shortening, which correlated with a narrower hypertrophic zone in their growth plates. Potential ADAMTS17 substrates identified by N-terminomics and yeast-2-hybrid screening revealed the ECM proteins fibronectin (FN1) and collagen VI (COL6A2). In primary ADAMTS10- and ADAMTS17-deficient skin fibroblasts, fibronectin deposition was impaired concomitant with aberrant intracellular accumulation of fibrillin-1. These findings support a role for ADAMTS17 in ECM protein secretion and assembly. Mechanistically, ADAMTS17 appears to be a critical regulator of ECM protein secretion or pericellular matrix assembly, whereas ADAMTS10 likely modulates ECM formation at later stages.
Combined ADAMTS10 and ADAMTS17 inactivation exacerbates bone shortening and compromises extracellular matrix formation.
Weill-Marchesani syndrome (WMS) is characterized by severe short stature, short hands and feet (brachydactyly), joint contractures, tight skin, and heart valve, eye, and skin anomalies. Whereas recessive WMS is caused by mutations in ADAMTS10, ADAMTS17, or LTBP2, dominant WMS is caused by mutations in FBN1 (encoding fibrillin-1). Since bone growth is driven by chondrocyte proliferation and hypertrophy in the growth plates, the genetics of WMS suggests that the affected ECM proteins act within the same pathway to regulate chondrocyte and growth plate function. Here, we investigated the role of the secreted ADAMTS proteases ADAMTS10 and ADAMTS17 in growth plate function and ECM formation. We generated Adamts10;Adamts17 double knockout (DKO) mice, which showed significant postnatal lethality compared to single Adamts10 or Adamts17 KO mice. Importantly, we observed severe bone shortening DKO mice, which correlated with a narrower hypertrophic zone in their growth plates. ADAMTS17 substrates identified by N-terminomics and yeast two-hybrid screening identified the ECM proteins fibronectin and collagen VI (COL6). However, validation experiments did not reveal direct proteolysis of either fibronectin or COL6 by ADAMTS17. We then investigated ECM formation in primary ADAMTS10- and ADAMTS17-deficient skin fibroblasts and observed compromised fibronectin deposition concomitant with aberrant intracellular accumulation of fibrillin-1. These findings support a role for ADAMTS17 in ECM protein secretion and assembly. Collectively, our data suggest that ADAMTS10 and ADAMTS17 regulate bone growth by regulating chondrocyte hypertrophy or hypertrophic chondrocyte turnover. Mechanistically, ADAMTS17 appears to be a critical regulator of ECM protein secretion or pericellular matrix assembly, whereas ADAMTS10 likely modulates ECM formation at later stages, possibly regulating the spatio-temporal deposition of fibrillin isoforms.
Publicações recentes
ADAMTS and ADAMTSL mutations in connective tissue disorders.
A novel homozygous ADAMTS10 frameshift variant in Weill-Marchesani syndrome in a Chinese family.
A novel compound heterozygous mutation in ADAMTS17 identified in a Chinese family with Weill-Marchesani syndrome.
A unique case of microspherophakia in adult Refsum disease.
Combined ADAMTS10 and ADAMTS17 inactivation exacerbates bone shortening and skin phenotypes.
📚 EuropePMC99 artigos no totalmostrando 71
A novel homozygous ADAMTS10 frameshift variant in Weill-Marchesani syndrome in a Chinese family.
BMC medical genomicsA novel compound heterozygous mutation in ADAMTS17 identified in a Chinese family with Weill-Marchesani syndrome.
International journal of ophthalmologyA unique case of microspherophakia in adult Refsum disease.
American journal of ophthalmology case reportsCombined ADAMTS10 and ADAMTS17 inactivation exacerbates bone shortening and skin phenotypes.
Life science allianceCombined ADAMTS10 and ADAMTS17 inactivation exacerbates bone shortening and compromises extracellular matrix formation.
bioRxiv : the preprint server for biologySecondary Angle Closure Glaucoma in Weill-Marchesani Syndrome.
Diagnostics (Basel, Switzerland)Fibrillin-1 Gene Variant p.Gly1754Ser Associated With Weill-Marchesani Syndrome Type 2: A Case Report.
CureusSuture dehiscence in patients with connective tissue disease: Marfan and Weill-Marchesani syndromes.
Archivos de la Sociedad Espanola de OftalmologiaCase Report: Two different acromelic dysplasia phenotypes in a Chinese family caused by a missense mutation in FBN1 and a literature review.
Frontiers in pediatricsGeleophysic dysplasia and Weill-Marchesani syndrome: ADAMTSL2 a possible common gene.
Ophthalmic genetics[Transscleral fixation of intraocular lens in the treatment of lens subluxation in children].
Vestnik oftalmologiiAutosomal Dominant Weill-Marchesani-Like Syndrome in a Chinese Family due to Novel Haplotypic Mutations in LTBP2.
Ophthalmic researchWeill-Marchesani syndrome: natural history and genotype-phenotype correlations from 18 news cases and review of literature.
Journal of medical geneticsMicrospherophakic Angle Closure Glaucoma in a Patient with Coffin-Siris Syndrome: Case Report.
The application of clinical geneticsCharacteristics and genotype-phenotype correlations in ADAMTS17 mutation-related Weill-Marchesani syndrome.
Experimental eye researchMarfan Syndrome: Enhanced Diagnostic Tools and Follow-up Management Strategies.
Diagnostics (Basel, Switzerland)A Novel Missense Mutation in the TGF-β-binding Protein-Like Domain 3 of FBN1 Causes Weill-Marchesani Syndrome with Intellectual Disability.
Advanced biomedical researchA Novel Mutation in the ADAMTS10 Associated with Weil-Marchesani Syndrome with a Unique Presentation of Developed Membranes Causing Severe Stenosis of the Supra Pulmonic, Supramitral, and Subaortic Areas in the Heart.
International journal of molecular sciencesNovel homozygous ADAMTS17 missense variant in Weill-Marchesani syndrome.
International journal of ophthalmologyFibrillin microfibril structure identifies long-range effects of inherited pathogenic mutations affecting a key regulatory latent TGFβ-binding site.
Nature structural & molecular biologyMicrospherophakia: A clinical approach and mini review with a case report.
Journal of family medicine and primary careAbnormal lens thickening in a child with Weill-Marchesani syndrome 4: A 3-year follow-up case report.
Frontiers in medicineCase report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome.
Frontiers in geneticsAdamts10 controls transforming growth factor β family signaling that contributes to retinal ganglion cell development.
Frontiers in molecular biosciencesADAMTS10 inhibits aggressiveness via JAK/STAT/c-MYC pathway and reprograms macrophage to create an anti-malignant microenvironment in gastric cancer.
Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer AssociationA Case of Refractory Childhood Glaucoma Secondary to Weill-Marchesani Syndrome: Management with Combined CO2 Laser-Assisted Sclerectomy Surgery and Trabeculectomy.
Chinese medical sciences journal = Chung-kuo i hsueh k'o hsueh tsa chihWeill-Marchesani syndrome 4 caused by compound heterozygosity of a maternal submicroscopic deletion and a paternal nonsense variant in the ADAMTS17 gene: A case report.
American journal of ophthalmology case reportsBilateral golden ring in the eye: Weill-Marchesani syndrome.
The National medical journal of IndiaPrimary scleral-fixated posterior chamber intraocular lenses in patients with congenital lens subluxation.
BMC ophthalmologyAltered Ocular Fibrillin Microfibril Composition in Mice With a Glaucoma-Causing Mutation of Adamts10.
Investigative ophthalmology & visual scienceWeill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature.
The American journal of case reportsA Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations.
Risk management and healthcare policyAlternative splicing of the metalloprotease ADAMTS17 spacer regulates secretion and modulates autoproteolytic activity.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologySecondary intraocular lens implantation using the flanged intrascleral fixation technique in pediatric aphakia: case series and review of literature.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusAcromelic dysplasias: how rare musculoskeletal disorders reveal biological functions of extracellular matrix proteins.
Annals of the New York Academy of SciencesThe quest for substrates and binding partners: A critical barrier for understanding the role of ADAMTS proteases in musculoskeletal development and disease.
Developmental dynamics : an official publication of the American Association of AnatomistsSutureless Scleral-Fixated Intraocular Lens Implantation for Refractive Rehabilitation in Eyes With Spherophakia.
Journal of vitreoretinal diseasesA novel pathogenic missense ADAMTS17 variant that impairs secretion causes Weill-Marchesani Syndrome with variably dysmorphic hand features.
Scientific reportsThe ADAMTS/Fibrillin Connection: Insights into the Biological Functions of ADAMTS10 and ADAMTS17 and Their Respective Sister Proteases.
Biomolecules[A case of secondary glaucoma in Weill-Marchesani syndrome].
Journal francais d'ophtalmologieA novel ADAMTS17 variant that causes Weill-Marchesani syndrome 4 alters fibrillin-1 and collagen type I deposition in the extracellular matrix.
Matrix biology : journal of the International Society for Matrix BiologyWeill-Marchesani Syndrome with Secondary Angle Closure Glaucoma.
Journal of glaucomaAdamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway.
Cellular and molecular life sciences : CMLSFibrin Glue-Assisted Intraocular Lens Fixation in Weill-Marchesani Syndrome.
Middle East African journal of ophthalmologyA novel nonsense mutation in ADAMTS17 caused autosomal recessive inheritance Weill-Marchesani syndrome from a Chinese family.
Journal of human genetics[Study of de novo point mutations in known genes among patients with unexplained intellectual disability or developmental delay].
Zhonghua yi xue za zhiAccommodative esotropia and Brown syndrome in a girl with recessive geleophysic dysplasia.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusDelineation of Novel Compound Heterozygous Variants in LTBP2 Associated with Juvenile Open Angle Glaucoma.
GenesThe RECK tumor-suppressor protein binds and stabilizes ADAMTS10.
Biology openFibrillin protein pleiotropy: Acromelic dysplasias.
Matrix biology : journal of the International Society for Matrix BiologyAdamts10 inactivation in mice leads to persistence of ocular microfibrils subsequent to reduced fibrillin-2 cleavage.
Matrix biology : journal of the International Society for Matrix BiologyADAMTS10-mediated tissue disruption in Weill-Marchesani syndrome.
Human molecular geneticsA report of three families with FBN1-related acromelic dysplasias and review of literature for genotype-phenotype correlation in geleophysic dysplasia.
European journal of medical geneticsCervical artery dissection expands the cardiovascular phenotype in FBN1-related Weill-Marchesani syndrome.
American journal of medical genetics. Part AUnusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease.
Scientific reportsADAMTS-10 and -6 differentially regulate cell-cell junctions and focal adhesions.
Scientific reportsIn vitro biometry of a human spherophakia.
Clinical & experimental optometryFBN1: The disease-causing gene for Marfan syndrome and other genetic disorders.
GeneMutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia.
Journal of medical geneticsAcute angle-closure glaucoma in a highly myopic patient secondary to Weill-Marchesani syndrome: histopathologic lens features.
International ophthalmologySkeletal manifestations of Marfan syndrome associated to heterozygous R2726W FBN1 variant: sibling case report and literature review.
BMC musculoskeletal disordersPotential Modifying Loci Associated With Primary Lens Luxation, Pedal Hyperkeratosis, and Ocular Phenotypes in Miniature Bull Terriers.
Investigative ophthalmology & visual scienceA new combined surgical approach in a patient with microspherophakia and developmental iridocorneal angle anomaly.
Nepalese journal of ophthalmology : a biannual peer-reviewed academic journal of the Nepal Ophthalmic Society : NEPJOPH[Multimodal imaging of angle closure secondary to spherophakia in Weill-Marchesani syndrome].
Journal francais d'ophtalmologieLensectomy, vitrectomy, and transvitreal ciliary body photocoagulation as primary treatment for glaucoma in microspherophakia.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusGolden ring in the eyes: Weill-Marchesani syndrome.
BMJ case reportsGlaucoma in iran and contributions of studies in iran to the understanding of the etiology of glaucoma.
Journal of ophthalmic & vision researchADAMTS proteins as modulators of microfibril formation and function.
Matrix biology : journal of the International Society for Matrix BiologyThe fibrillin microfibril scaffold: A niche for growth factors and mechanosensation?
Matrix biology : journal of the International Society for Matrix BiologyExtended-depth spectral-domain optical coherence tomography imaging of the crystalline lens in Weill-Marchesani-like syndrome.
JCRS online case reportsWeill-Marchesani syndrome with advanced glaucoma and corneal endothelial dysfunction: a case report and literature review.
BMC ophthalmologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Weill-Marchesani.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Weill-Marchesani
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A novel homozygous ADAMTS10 frameshift variant in Weill-Marchesani syndrome in a Chinese family.
- A novel compound heterozygous mutation in ADAMTS17 identified in a Chinese family with Weill-Marchesani syndrome.
- A unique case of microspherophakia in adult Refsum disease.
- Combined ADAMTS10 and ADAMTS17 inactivation exacerbates bone shortening and skin phenotypes.
- Combined ADAMTS10 and ADAMTS17 inactivation exacerbates bone shortening and compromises extracellular matrix formation.
- ADAMTS and ADAMTSL mutations in connective tissue disorders.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3449(Orphanet)
- MONDO:0018096(MONDO)
- GARD:4936(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3961695(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
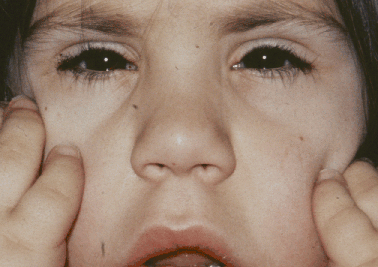
Síndrome Weill-Marchesani
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata