A Diabetes e Surdez Hereditários da Mãe (MIDD) é uma doença relacionada às mitocôndrias, que se manifesta como diabetes transmitido da mãe para o filho e um tipo de surdez que afeta os nervos da audição.
Introdução
O que você precisa saber de cara
A Diabetes e Surdez Hereditários da Mãe (MIDD) é uma doença relacionada às mitocôndrias, que se manifesta como diabetes transmitido da mãe para o filho e um tipo de surdez que afeta os nervos da audição.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 5 sintomas em outras categorias
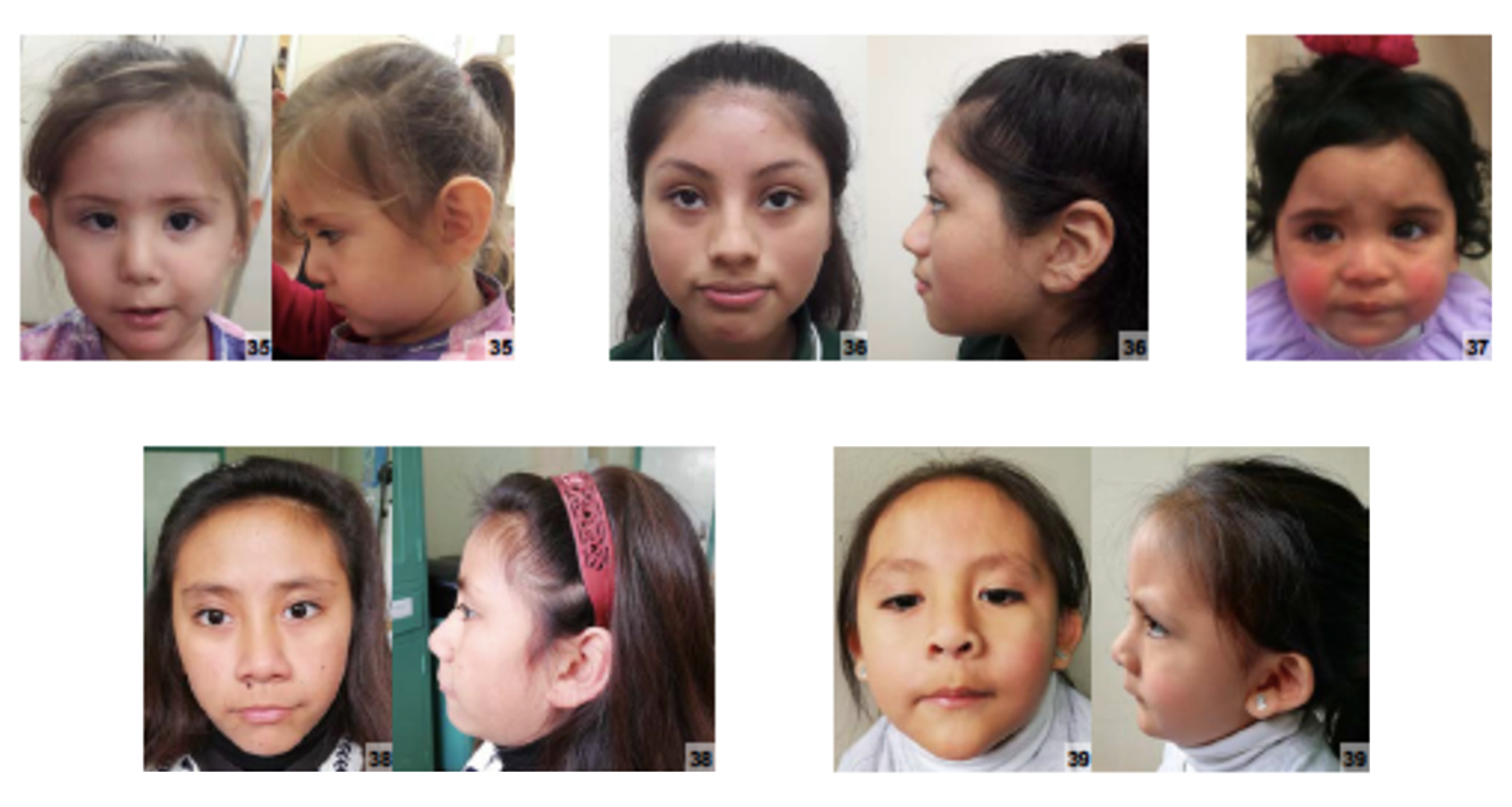
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 15 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Diabetes e surdez com hereditariedade materna
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Mostrando amostra de 2 publicações de um total de 130
Unraveling the genetic spectrum of inherited deaf-blindness in Portugal.
Syndromic genetic disorders affecting vision can also cause hearing loss, and Usher syndrome is by far the most common etiology. However, many other conditions can present dual sensory impairment. Accurate diagnosis is essential for providing patients with genetic counseling, prognostic information, and appropriate resources. This study aimed to describe the genetic profile of combined inherited deaf-blindness in Portugal. This was a cross-sectional study conducted at a tertiary hospital in Portugal. Patients were identified through the national, web-based inherited retinal dystrophies registry (IRD-PT, retina.com.pt). Demographics, clinical, and genetic data were retrieved from individual patient records. Genetic variants were classified according to the American College of Medical Genetics and Genomics; only likely pathogenic or pathogenic variants were considered relevant for solved cases. Eighty-four patients (58.3% males; mean age 40.0 ± 17.9 years) from 71 families were included. Usher syndrome was the most frequent etiology (71.4%) followed by Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract syndrome (6.0%), Autosomal dominant optic atrophy plus (4.8%) and cone-rod dystrophy and hearing loss (4.8%). Other less frequent etiologies included Alport syndrome (2.4%), Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (2.4%), Heimler syndrome (2.4%), Senior-Loken syndrome (1.2%), Waardenburg syndrome (1.2%), Maternally inherited diabetes and deafness (1.2%), and Stickler syndrome (1.2%). The overall diagnostic yield of deleterious variants in our deaf-blind cohort was 73.2%. A total of 55 genetic variants were identified across 16 different genes; 11 of these variants are novel and herein reported for the first time. This is the first study to describe the genetic profile of patients with dual sensory impairment in Portugal, highlighting the genetic heterogeneity associated with inherited deaf-blindness. Usher syndrome was the most prevalent cause in this cohort. Nevertheless, several other less frequent causes must also be considered. This study showed a high diagnostic yield and reported 11 novel genetic variants, thereby contributing to expand the mutational spectrum of these conditions.
Low prevalence of patients with mitochondrial disease in the German/Austrian DPV diabetes registry.
The aim of this study was to characterize the phenotype and treatment of young patients (manifestation <30 years) with diabetes of mitochondrial origin (DMO), based on the German/Austrian DPV (Diabetes Patienten Verlaufsdokumentation) registry. Only 13 (0.02 %) of all patients with diabetes in this cohort were identified with DMO, mainly due to the Kearns-Sayre (n = 5), Pearson (n = 3), or mitochondrial myopathy, encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome (n = 2). The onset of DMO (14.2, interquartile range (IQR) 7.1-16 years) was later than diabetes onset in individuals with T1D but earlier than in T2D. At manifestation, patients exhibited a mild elevation of blood glucose concentrations (251, IQR 178-299 mg/dl) without ketoacidosis. They had lower body mass index (BMI) values (-1.39 ± 0.28 kg/m(2)) than peers with T1D or T2D (p < 0.0001) and higher triglycerides (211, IQR 134-574 mg/dl) than in T1D (p = 0.04) while there was a high rate of dyslipidemia (86 %). Insulin requirements (0.58, IQR 0.37-0.90 U/kg/d) were between T1D and T2D while glucometabolic control (glycated hemoglobin A1c (HbA1c) 7.4 ± 0.52 %) in DMO was comparable to age-matched T2D and stable over a 5-year follow-up. Primary mitochondrial disorders are a rare cause of juvenile diabetes and likely to be underdiagnosed. As there is clinical overlap with T1D and T2D, dyslipidemia and low body weight may help to identify further DMO cases. • In adults diabetes of mitochondrial origin (DMO) is a rare cause of non-autoimmune diabetes, affecting about 0.8 % of diabetes cases. • Common features are a maternal family history of diabetes, hearing loss and neurological abnormalities. What is New: • In our juvenile cohort 0.02 % of diabetes patients (age < 30 years) were affected by DMO, while Kearns Sayre, MELAS and Pearson syndrome were the most frequent entities. • Juvenile DMO patients exhibited dyslipidemia, higher triglycerides and a lower BMI than peers with T1D or T2D, while some patients also showed retinal changes.
Publicações recentes
COQ8A-related Primary Coenzyme Q10 Deficiency Mimicking Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Syndrome: A Pediatric Case Report and Review of Mitochondrial Mimics.
Clinical Validation of Digital PCR for Precise m.3243A>G Heteroplasmy Quantification in Early-Onset Diabetes.
A heterozygous pathogenic RPE65 variant phenocopies a mitochondrial retinopathy.
📖 RevisãoScreening for Maternally Inherited Diabetes and Deafness in Large Cohorts of Hearing Impaired and Diabetic Patients.
Biomarking MELAS with neurofilament light chain and circulating cell free mitochondrial DNA.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Diabetes e surdez com hereditariedade materna.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Diabetes e surdez com hereditariedade materna
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Unraveling the genetic spectrum of inherited deaf-blindness in Portugal.
- Low prevalence of patients with mitochondrial disease in the German/Austrian DPV diabetes registry.
- COQ8A-related Primary Coenzyme Q10 Deficiency Mimicking Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Syndrome: A Pediatric Case Report and Review of Mitochondrial Mimics.
- Clinical Validation of Digital PCR for Precise m.3243A>G Heteroplasmy Quantification in Early-Onset Diabetes.
- A heterozygous pathogenic RPE65 variant phenocopies a mitochondrial retinopathy.
- Screening for Maternally Inherited Diabetes and Deafness in Large Cohorts of Hearing Impaired and Diabetic Patients.
- Biomarking MELAS with neurofilament light chain and circulating cell free mitochondrial DNA.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:225(Orphanet)
- OMIM OMIM:520000(OMIM)
- MONDO:0010785(MONDO)
- GARD:4003(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q4313947(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Diabetes e surdez com hereditariedade materna
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata