A encefalopatia causada pela deficiência da enzima sulfito oxidase é uma condição rara que afeta o cérebro e o metabolismo do corpo. Ela se manifesta por convulsões, um problema cerebral que piora progressivamente e o deslocamento do cristalino (a lente natural do olho).
Introdução
O que você precisa saber de cara
A encefalopatia causada pela deficiência da enzima sulfito oxidase é uma condição rara que afeta o cérebro e o metabolismo do corpo. Ela se manifesta por convulsões, um problema cerebral que piora progressivamente e o deslocamento do cristalino (a lente natural do olho).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 31 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 95 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
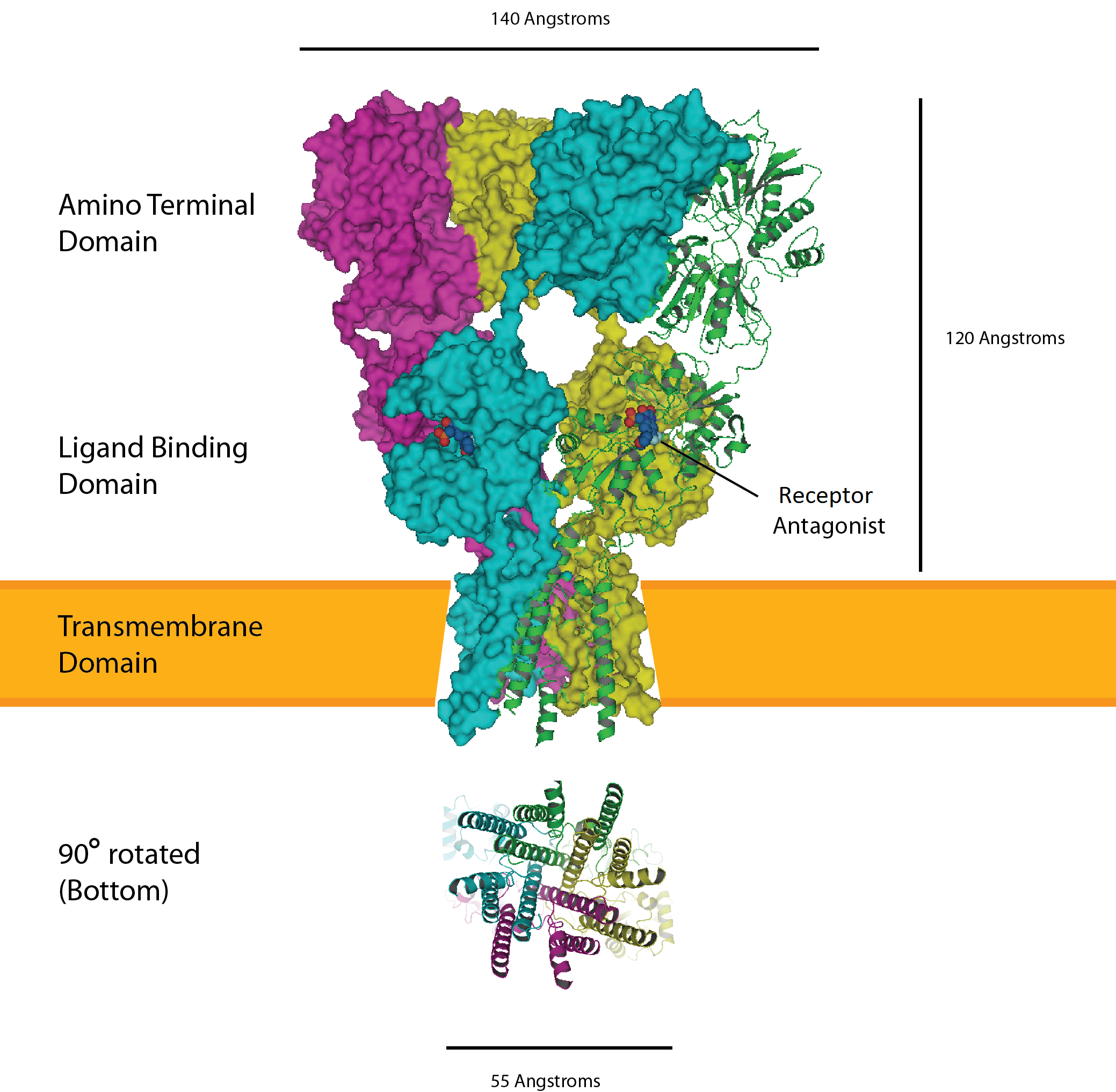
Catalyzes the oxidation of sulfite to sulfate, the terminal reaction in the oxidative degradation of sulfur-containing amino acids
Mitochondrion intermembrane space
Sulfite oxidase deficiency, isolated
A life-threatening, autosomal recessive neurometabolic disorder characterized by severe neurological impairment. Classic ISOD manifests in the first few hours to days of life and is characterized by intractable seizures, feeding difficulties, rapidly progressive encephalopathy, microcephaly, and profound intellectual disability. Children usually die during the first few months of life. Mild ISOD manifests in infancy or early childhood and is characterized by ectopia lentis that is variably present, developmental delay and regression, movement disorder characterized by dystonia and choreoathetosis, ataxia, and rarely acute hemiplegia due to metabolic stroke.
Catalytic subunit of the molybdopterin synthase complex, a complex that catalyzes the conversion of precursor Z into molybdopterin. Acts by mediating the incorporation of 2 sulfur atoms from thiocarboxylated MOCS2A into precursor Z to generate a dithiolene group (By similarity) (PubMed:12732628, PubMed:15073332, PubMed:25709896). Together with MBIP, inhibits the activity of stress kinase EIF2AK2/PKR; this may suppress JNK activation and subsequent stress-responsive transcription, or suppress eIF
Cytoplasm, cytosolNucleus
Molybdenum cofactor deficiency B
An autosomal recessive metabolic disorder characterized by neonatal onset of intractable seizures, opisthotonus, and facial dysmorphism associated with hypouricemia and elevated urinary sulfite levels. Affected individuals show severe neurologic damage and often die in early childhood.
Isoform MOCS1A and isoform MOCS1B probably form a complex that catalyzes the conversion of 5'-GTP to cyclic pyranopterin monophosphate (cPMP) (PubMed:11891227, PubMed:23627491, PubMed:29368224, PubMed:31996372). MOCS1A catalyzes the cyclization of GTP to (8S)-3',8-cyclo-7,8-dihydroguanosine 5'-triphosphate and MOCS1B catalyzes the subsequent conversion of (8S)-3',8-cyclo-7,8-dihydroguanosine 5'-triphosphate to cPMP (PubMed:11891227, PubMed:23627491, PubMed:29368224, PubMed:31996372) Has very wea
Mitochondrion matrixCytoplasm, cytosolCytoplasm
Molybdenum cofactor deficiency A
An autosomal recessive metabolic disorder leading to the pleiotropic loss of molybdoenzyme activities. It is clinically characterized by onset in infancy of poor feeding, intractable seizures, severe psychomotor retardation, and death in early childhood in most patients.
Microtubule-associated protein involved in membrane protein-cytoskeleton interactions. It is thought to anchor the inhibitory glycine receptor (GLYR) to subsynaptic microtubules (By similarity). Acts as a major instructive molecule at inhibitory synapses, where it also clusters GABA type A receptors (PubMed:25025157, PubMed:26613940) Also has a catalytic activity and catalyzes two steps in the biosynthesis of the molybdenum cofactor. In the first step, molybdopterin is adenylated. Subsequently,
Postsynaptic cell membraneCell membraneCytoplasm, cytosolCytoplasm, cytoskeletonCell projection, dendritePostsynaptic density
Molybdenum cofactor deficiency C
A form of molybdenum cofactor deficiency, an autosomal recessive metabolic disorder leading to the pleiotropic loss of molybdoenzyme activities. It is clinically characterized by onset in infancy of poor feeding, intractable seizures, severe psychomotor retardation, and death in early childhood in most patients.
Variantes genéticas (ClinVar)
310 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Encefalopatia devido à deficiência de sulfito oxidase
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Ultra-orphan diseases: A cross-sectional quantitative analysis of the natural history of isolated sulfite oxidase deficiency.
Isolated sulfite oxidase deficiency (ISOD; OMIM #272300) is a devastating rare neurometabolic disorder due to biallelic pathogenic variants in the SUOX gene, that typically results in neonatal refractory epilepsy and progressive severe encephalopathy. Knowledge on the quantitative natural history of ISOD is limited and clinical outcome parameters for future clinical trials remain to be defined. We performed a comprehensive analysis of published cases (N=74) with ISOD applying quantitative retrospective natural history modeling (QUARNAM). Main outcome parameters were age of disease onset, diagnostic delay and survival. Clinical characteristics and potential associations between biochemical parameters and clinical outcome (i.e. age of disease onset, survival) were explored. The median survival period of the study cohort was 60 months. ISOD typically presented shortly after birth with a median age of onset of 3 days. Median age at diagnosis was 10 months, leading to a substantial median diagnostic delay of 5.7 months. Homocysteine concentrations in plasma correlated with age of disease onset. An association of biochemical parameters of cysteine metabolism and survival could not be identified. The present analysis describes long-term outcome measures adding to the quantitative understanding of the natural history of ISOD, which might be helpful in the planning of prospective clinical trials and potentially stimulate development of targeted therapies in the future.
Early postnatal hepatocyte transplantation in a child with molybdenum cofactor deficiency type B.
Molybdenum cofactor deficiencies (MoCD) are a group of inborn errors of metabolism that result in impaired synthesis of molybdenum cofactor, crucial for the function of three oxidases (sulfite oxidase, xanthine oxidase and aldehyde oxidase). Most patients present with severe neonatal-onset epileptic encephalopathy, hypotonia, poor feeding and apnoea, with death typically occurring within the first three years of life. Whilst there is now an emerging therapy for MoCD Type A (cPMP/fosdenopterin), this treatment is not effective for MoCD Type B and there is no treatment for isolated sulfite oxidase deficiency (ISOD). Liver directed gene delivery is a potential alternative therapy for sulfite intoxication disorders. We report an attempt to use hepatocyte transplantation as a treatment option for MoCD Type B, in an infant with a strong family history of neonatal-onset disease and early mortality. Six transfusions of hepatocytes were given between Day 1 and Day 18 of life, totalling around 1 × 109 cells with immunosuppressive cover. Concomitantly dietary protein restriction was maintained at 2 g/kg, including 0.7 g/kg of methionine- and cyst(e)ine-free amino acid mixture. The aim was to utilize hepatocyte transplantation as a bridge to liver transplantation. Whilst there was evidence of biochemical stabilization with reduction in concentrations of sulfite and S-sulfocysteine and a moderate increase in urate levels compared to the sibling, the treatment was not able to prevent acute brain injury from sulfite toxicity which was evident in neuroimaging at 35 h of age. This correlated clinically with ongoing seizures as well as minimal developmental progress.
Increased ROS levels, antioxidant defense disturbances and bioenergetic disruption induced by thiosulfate administration in the brain of neonatal rats.
Sulfite oxidase deficiencies, either caused by deficiency of the apoenzyme or the molybdenum cofactor, and ethylmalonic encephalopathy are inherited disorders that impact sulfur metabolism. These patients present with severe neurodeterioration accompanied by cerebral cortex and cerebellum abnormalities, and high thiosulfate levels in plasma and tissues, including the brain. We aimed to clarify the mechanisms of such abnormalities, so we assessed the ex vivo effects of thiosulfate administration on energetic status and oxidative stress markers in cortical and cerebellar tissues of newborn rats. Thiosulfate (0.5 µmol/g) or PBS (vehicle) was injected into the fourth ventricle of rat pups. Thirty minutes after the injection, animals were euthanized and the brain structures were utilized for the experiments. Our data showed that thiosulfate decreased the reduced glutathione (GSH) concentrations, and superoxide dismutase (SOD), catalase (CAT) and glutathione S-transferase (GST) activities in the cortical structure. Thiosulfate also increased DCFH oxidation, hydrogen peroxide generation and glutathione reductase activity. In the cerebellum, thiosulfate reduced SOD and glutathione peroxidase activities but increased GST and CAT activities as well as DCFH oxidation. Regarding energy metabolism, thiosulfate specifically decreased complex IV activity in the cortex, whereas it increased cerebellar complex I and creatine kinase activities, indicating bioenergetic disturbances. The results suggest that the accumulation of thiosulfate causing redox disruption and bioenergetic alterations has a prominent role in the pathogenesis of sulfur metabolism deficiencies.
Timing of cerebral damage in molybdenum cofactor deficiency: A meta-analysis of case reports.
Molybdenum cofactor deficiency (MoCD) classically presents shortly after birth, with neurological symptoms ascribed to postnatal toxicity of accumulating sulphite. Case reports suggest that cerebral damage associated with MoCD may have a prenatal onset. A meta-analysis of case reports was performed on individuals with genetically proven MoCD retrieved through a systematic review and in-house search. Cases were categorized as classical or late-onset, based on the time of onset of symptoms. Available cerebral images were scored for the presence of restricted diffusion, pathological signal, subcortical cysts, and atrophy. Estimated onset of each event and the minimal number of events needed to explain the observed imaging abnormalities were deduced by combining age at imaging, type of imaging abnormality, and known natural evolution of the imaging abnormalities. Of a total of 30 retrieved cases, 21 were classical. Prenatal origin of damage was possible in all classical cases and certain in 11 of 21 (52%). Multiple events were deduced in 5/21 classical cases based on imaging data alone and in 11 of 21 cases when presuming that a postnatal onset of symptoms signifies a recent event. Multiple, but postnatal, events were also described in 3 of 9 late-onset cases. Prenatal onset of cerebral damage in patients with classical MoCD is more frequently encountered than anticipated. It may have been overlooked by the overwhelming postnatal symptoms erroneously pointing to a single culprit. This insight is important when counseling for prognosis, particularly in the context of considering the timing and anticipated prospects of therapeutic intervention.
Consensus guidelines for the diagnosis and management of isolated sulfite oxidase deficiency and molybdenum cofactor deficiencies.
Sulfite intoxication is the hallmark of four ultrarare disorders that are caused by impaired sulfite oxidase activity due to genetic defects in the synthesis of the molybdenum cofactor or of the apoenzyme sulfite oxidase. Delays on the diagnosis of these disorders are common and have been caused by their unspecific presentation of acute neonatal encephalopathy with high early mortality, followed by the evolution of dystonic cerebral palsy and also by the lack of easily available and reliable diagnostic tests. There is significant variation in survival and in the quality of symptomatic management of affected children. One of the four disorders, molybdenum cofactor deficiency type A (MoCD-A) has recently become amenable to causal treatment with synthetic cPMP (fosdenopterin). The evidence base for the rational use of cPMP is very limited. This prompted the formulation of these clinical guidelines to facilitate diagnosis and support the management of patients. The guidelines were developed by experts in diagnosis and treatment of sulfite intoxication disorders. It reflects expert consensus opinion and evidence from a systematic literature search.
Publicações recentes
Developing an explainable machine learning model to predict false-negative citrin deficiency cases in newborn screening.
5-Year Results From the AMPLATZER Amulet Left Atrial Appendage Occluder Randomized Controlled Trial.
🥇 Ensaio randomizadoCost-of-illness studies of inherited retinal diseases: a systematic review.
🥇 Revisão sistemáticaTherapeutic effects of extracorporeal shock wave therapy on patients with spastic cerebral palsy and Rett syndrome: clinical and ultrasonographic findings.
Craniofacial growth and function in achondroplasia: a multimodal 3D study on 15 patients.
📚 EuropePMCmostrando 26
Ultra-orphan diseases: A cross-sectional quantitative analysis of the natural history of isolated sulfite oxidase deficiency.
PloS oneEarly postnatal hepatocyte transplantation in a child with molybdenum cofactor deficiency type B.
Molecular genetics and metabolismIncreased ROS levels, antioxidant defense disturbances and bioenergetic disruption induced by thiosulfate administration in the brain of neonatal rats.
Metabolic brain diseaseTiming of cerebral damage in molybdenum cofactor deficiency: A meta-analysis of case reports.
Genetics in medicine openA case report of MoCD etiology in a neonate: A novel homozygous MoCS2 variant.
Clinical case reportsConsensus guidelines for the diagnosis and management of isolated sulfite oxidase deficiency and molybdenum cofactor deficiencies.
Journal of inherited metabolic diseaseEpilepsy in sulfite oxidase deficiency and related disorders: insights from neuroimaging and genetics.
Epilepsy & behavior : E&BSulfite Impairs Bioenergetics and Redox Status in Neonatal Rat Brain: Insights into the Early Neuropathophysiology of Isolated Sulfite Oxidase and Molybdenum Cofactor Deficiencies.
Cellular and molecular neurobiologyWhole exome sequencing identified a homozygous novel mutation in SUOX gene causes extremely rare autosomal recessive isolated sulfite oxidase deficiency.
Clinica chimica acta; international journal of clinical chemistryCase Report: Electroencephalography in a neonate with isolated sulfite oxidase deficiency - a case report and literature review.
HRB open researchSevere isolated sulfide oxidase deficiency with a novel mutation.
The Turkish journal of pediatricsMachine learning-based identification and characterization of 15 novel pathogenic SUOX missense mutations.
Molecular genetics and metabolismPostmortem whole-genome sequencing on a dried blood spot identifies a novel homozygous SUOX variant causing isolated sulfite oxidase deficiency.
Cold Spring Harbor molecular case studiesNovel Compound Heterozygous Pathogenic Variants in SUOX Cause Isolated Sulfite Oxidase Deficiency in a Chinese Han Family.
Frontiers in geneticsVery early neuroimages of sulfite oxidase deficiency mimicing severe hypoxic ischemic encephalopathy in a neonate.
Pediatrics and neonatologyLessons Learned from Inherited Metabolic Disorders of Sulfur-Containing Amino Acids Metabolism.
The Journal of nutritionTeaching NeuroImages: Early imaging of sulfite oxidase deficiency mimics severe hypoxic ischemic encephalopathy.
NeurologyMolybdenum Cofactor Deficiency: Mega Cisterna Magna in Two Consecutive Pregnancies and Review of the Literature.
The application of clinical geneticsThe Role of Oxidative Stress and Bioenergetic Dysfunction in Sulfite Oxidase Deficiency: Insights from Animal Models.
Neurotoxicity researchNeonatal epilepsies: Clinical management.
Seminars in fetal & neonatal medicineIsolated sulfite oxidase deficiency.
Journal of inherited metabolic diseasePrenatal brain disruption in isolated sulfite oxidase deficiency.
Orphanet journal of rare diseasesBezafibrate prevents mitochondrial dysfunction, antioxidant system disturbance, glial reactivity and neuronal damage induced by sulfite administration in striatum of rats: Implications for a possible therapeutic strategy for sulfite oxidase deficiency.
Biochimica et biophysica acta. Molecular basis of diseaseMolybdenum cofactor and isolated sulphite oxidase deficiencies: Clinical and molecular spectrum among Egyptian patients.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society[Sulfite oxidase activity deficiency caused by cofactor molybdenum deficiency: A case of early severe encephalopathy].
Archives de pediatrie : organe officiel de la Societe francaise de pediatrieClinical and neuroimaging features as diagnostic guides in neonatal neurology diseases with cerebellar involvement.
Cerebellum & ataxiasAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Encefalopatia devido à deficiência de sulfito oxidase.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Encefalopatia devido à deficiência de sulfito oxidase
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Ultra-orphan diseases: A cross-sectional quantitative analysis of the natural history of isolated sulfite oxidase deficiency.
- Early postnatal hepatocyte transplantation in a child with molybdenum cofactor deficiency type B.
- Increased ROS levels, antioxidant defense disturbances and bioenergetic disruption induced by thiosulfate administration in the brain of neonatal rats.
- Timing of cerebral damage in molybdenum cofactor deficiency: A meta-analysis of case reports.
- Consensus guidelines for the diagnosis and management of isolated sulfite oxidase deficiency and molybdenum cofactor deficiencies.
- Developing an explainable machine learning model to predict false-negative citrin deficiency cases in newborn screening.
- 5-Year Results From the AMPLATZER Amulet Left Atrial Appendage Occluder Randomized Controlled Trial.
- Cost-of-illness studies of inherited retinal diseases: a systematic review.
- Therapeutic effects of extracorporeal shock wave therapy on patients with spastic cerebral palsy and Rett syndrome: clinical and ultrasonographic findings.
- Craniofacial growth and function in achondroplasia: a multimodal 3D study on 15 patients.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:833(Orphanet)
- MONDO:0019358(MONDO)
- GARD:16549(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56014289(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Encefalopatia devido à deficiência de sulfito oxidase
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata