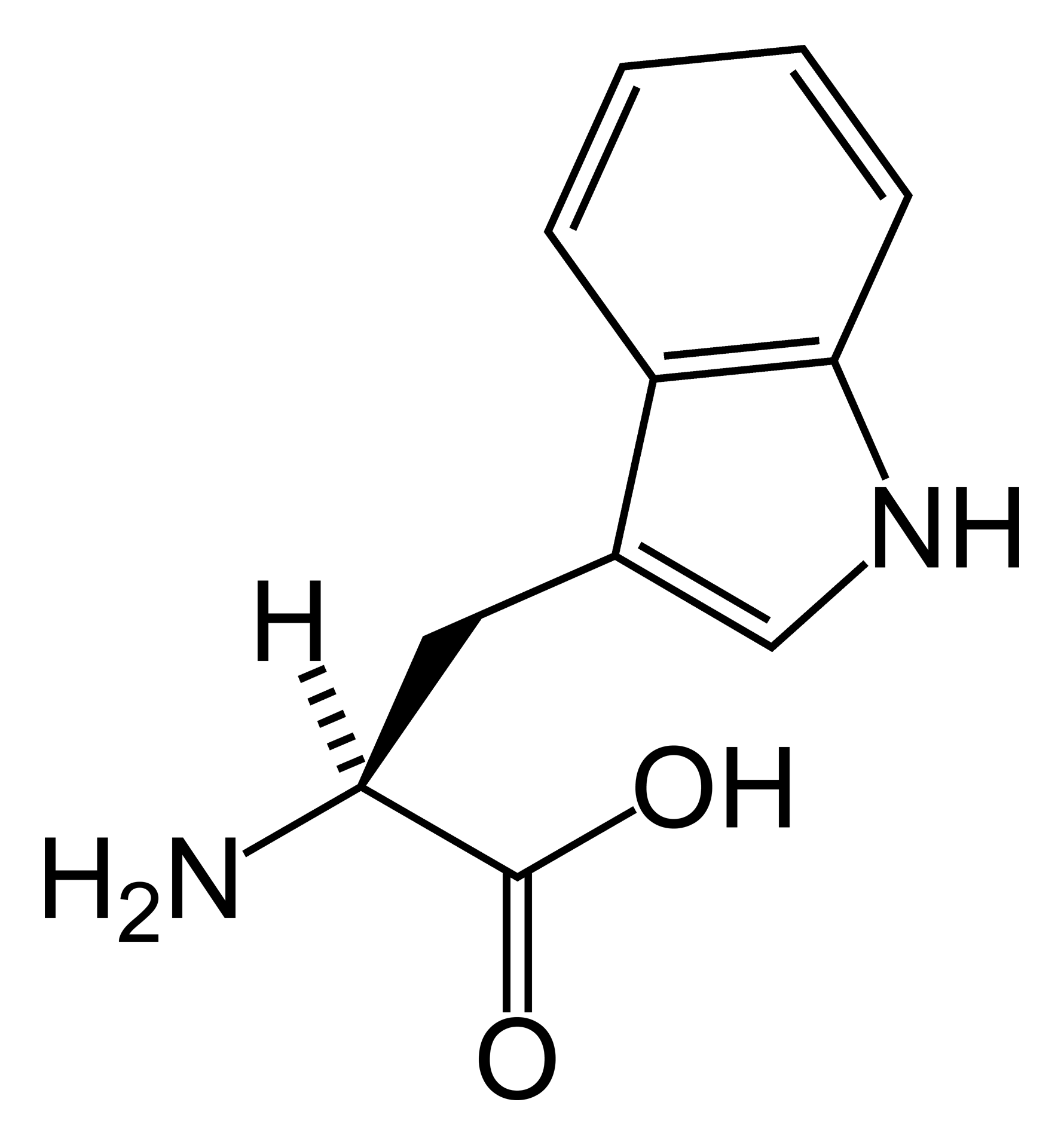
A doença de Hartnup é uma condição rara do metabolismo (que afeta como o corpo processa certas substâncias). Ela se enquadra em um grupo de problemas onde há um transporte inadequado de aminoácidos neutros — nutrientes essenciais — tanto nos rins quanto no intestino. Alguns desses aminoácidos são: triptofano, alanina, asparagina, glutamina, histidina, isoleucina, leucina, fenilalanina, serina, treonina, tirosina e valina.
Introdução
O que você precisa saber de cara
A doença de Hartnup é uma condição rara do metabolismo (que afeta como o corpo processa certas substâncias). Ela se enquadra em um grupo de problemas onde há um transporte inadequado de aminoácidos neutros — nutrientes essenciais — tanto nos rins quanto no intestino. Alguns desses aminoácidos são: triptofano, alanina, asparagina, glutamina, histidina, isoleucina, leucina, fenilalanina, serina, treonina, tirosina e valina.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 40 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisTransporter that mediates resorption of neutral amino acids across the apical membrane of renal and intestinal epithelial cells (PubMed:15286787, PubMed:15286788, PubMed:18424768, PubMed:18484095, PubMed:19185582, PubMed:26240152). This uptake is sodium-dependent and chloride-independent (PubMed:15286787, PubMed:15286788, PubMed:19185582). Requires CLTRN in kidney or ACE2 in intestine for cell surface expression and amino acid transporter activity (PubMed:18424768, PubMed:19185582)
Cell membraneApical cell membrane
Hartnup disorder
Autosomal recessive abnormality of renal and gastrointestinal neutral amino acid transport noted for its clinical variability. First described in 1956, HND is characterized by increases in the urinary and intestinal excretion of neutral amino acids. Individuals with typical Hartnup aminoaciduria may be asymptomatic, some develop a photosensitive pellagra-like rash, attacks of cerebellar ataxia and other neurological or psychiatric features. Although the definition of HND was originally based on clinical and biochemical abnormalities, its marked clinical heterogeneity has led to it being known as a disorder with a consistent pathognomonic neutral hyperaminoaciduria.
Plays an important role in amino acid transport by acting as binding partner of amino acid transporters SLC6A18 and SLC6A19, regulating their trafficking on the cell surface and their amino acid transporter activity (By similarity). May also play a role in trafficking of amino acid transporters SLC3A1 and SLC7A9 to the renal cortical cell membrane (By similarity). Regulator of SNARE complex function (PubMed:16330323). Stimulator of beta cell replication (PubMed:16330323)
Cell membrane
Variantes genéticas (ClinVar)
365 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Hartnup
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Dermatology-related disorders named after patients.
This contribution presents 14 dermatology-related eponyms that honor the patients who helped define the clinical entities and disorders that bear their names or initials. These clinical entities and disorders include: (1) anti-La antibodies, (2) anti-Ro antibodies, (3) anti-Sm antibodies, (4) B-K mole syndrome, (5) Hela cells, (6) Carrion disease, (7) Christmas disease, (8) Cowden syndrome, (9) Hageman disease, (10) Hartnup disease, (11) Mortimer malady, (12) rickettsiosis, (13) Schamroth sign, and (14) Trousseau syndrome.
Hartnup disease-causing SLC6A19 mutations lead to B0AT1 aberrant trafficking and ACE2 mis-localisation implicating the endoplasmic reticulum protein quality control.
The interaction between angiotensin-converting enzyme 2 (ACE2) and the sodium-dependent Broad neutral Amino acid Transporter 1 (B0AT1), encoded by the SLC6A19 gene, is increasingly recognized as pivotal in both physiological and pathological contexts. B0AT1 facilitates neutral amino acid transport and nutrient absorption, while ACE2 regulates vascular homeostasis and inflammation through the renin-angiotensin system. Mutations in SLC6A19 are implicated in Hartnup disease, a metabolic disorder characterized by defective amino acid transport. However, the cellular mechanisms underlying Hartnup disease-causing mutations' impact on B0AT1 and ACE2 function remain unclear. This study evaluated the subcellular localization and trafficking of 18 Hartnup disease-causing B0AT1 variants using experimental approaches including biochemical assays and In Silico analysis. The impact of these variants on ACE2 trafficking and plasma membrane targeting was also assessed to elucidate their interplay. Nine B0AT1 variants (R57C, G93R, R95P, R178Q, L242P, G284R, S303L, D517G, P579L) were found to be retained in the endoplasmic reticulum, impairing their trafficking to the plasma membrane. These variants were distributed across multiple B0AT1 structural domains. Importantly, several of these ER-retained variants, particularly R178Q and S303L, significantly disrupted ACE2 intracellular trafficking and its localization to the plasma membrane, indicating a direct effect on ACE2 subcellular targeting. The findings reveal that Hartnup disease-causing mutations can lead to ER retention of B0AT1, which in turn has a variable effect on ACE2 trafficking. This disruption likely contributes to Hartnup disease pathogenesis by impairing amino acid transport and may influence ACE2-mediated physiological functions beyond the renin-angiotensin system. Understanding these molecular mechanisms enhances insight into ACE2-B0AT1 interactions and could inform future therapeutic strategies and biomarker development for related disorders. Further research is needed to explore these pathways and their implications in disease.
Dermatology-related disorders named after patients.
This contribution presents 14 dermatology-related eponyms that honor the patients who helped to define the clinical entities and disorders that bear their names or initials. These clinical entities and disorders include: (1) anti-La antibodies, (2) anti-Ro antibodies, (3) anti-Sm antibodies, (4) B-K mole syndrome, (5) HeLa cells, (6) Carrion disease, (7) Christmas disease, (8) Cowden syndrome, (9) Hageman disease, (10) Hartnup disease, (11) Mortimer malady, (12) rickettsiosis, (13) Schamroth sign, and (14) Trousseau syndrome.
Hartnup Disease-Associated Psychosis: A Dwindling Phenomenon or Just Underreported?
Adult Neuropsychiatric Manifestation of Hartnup Disease With a Novel SLCA6A19 Variant: A Case Report.
In adults, inborn metabolic diseases are often missed in routine diagnostic settings due to a low level of suspicion. A patient in their twenties was admitted for an apparent acute exacerbation of anxiety disorder. Medical treatment was unsuccessful, and presumed catatonic psychosis was treated by electroconvulsive treatment. The patient was referred to neurology with reduced level of consciousness, mutism with no targeted movements, obvious anxiety and tetraspasticity, eczema, and reduced body weight. EEG was normal; repeat brain MRI showed progressive atrophy and leukoencephalopathy. Autoimmune encephalitis was assumed and treated with plasma exchange, high-dose glucocorticoids, and intravenous immunoglobulin. Repeated CSF analyses remained normal. Metabolic workup showed hyperaminociduria, low neutral amino acids, and undetectable tryptophane. Whole-exome sequencing and segregation analysis revealed compound heterozygous, pathogenic and a novel, likely pathogenic variant in the SLC6A19 gene: c.718C>T, p.(Arg240*) and c.170G>A, p.(Arg57His). Diagnosing Hartnup disease, high-protein diet, and niacin supplementation led to rapid considerable improvement. At 4 months, plasma amino acids were normal; communication and behavior were age-adequate; and spasticity had almost resolved, but polyneuropathy was unchanged. Metabolic workup and whole-exome sequencing are recommended in rapidly progressive neuropsychiatric disease, especially with additional neurologic signs and when standard treatment fails.
Publicações recentes
Dermatology-related disorders named after patients.
📖 RevisãoDermatology-related disorders named after patients.
Hartnup disease-causing SLC6A19 mutations lead to B0AT1 aberrant trafficking and ACE2 mis-localisation implicating the endoplasmic reticulum protein quality control.
Hartnup Disease-Associated Psychosis: A Dwindling Phenomenon or Just Underreported?
Adult Neuropsychiatric Manifestation of Hartnup Disease With a Novel SLCA6A19 Variant: A Case Report.
📚 EuropePMC85 artigos no totalmostrando 16
Dermatology-related disorders named after patients.
Clinics in dermatologyHartnup disease-causing SLC6A19 mutations lead to B0AT1 aberrant trafficking and ACE2 mis-localisation implicating the endoplasmic reticulum protein quality control.
Frontiers in cell and developmental biologyHartnup Disease-Associated Psychosis: A Dwindling Phenomenon or Just Underreported?
Journal of the Academy of Consultation-Liaison PsychiatryAdult Neuropsychiatric Manifestation of Hartnup Disease With a Novel SLCA6A19 Variant: A Case Report.
Neurology. GeneticsNew aspects for the brain in Hartnup disease based on mining of high-resolution cellular mRNA expression data for SLC6A19.
IBRO neuroscience reportsEpisodic Ataxias: Primary and Secondary Etiologies, Treatment, and Classification Approaches.
Tremor and other hyperkinetic movements (New York, N.Y.)Systemic tryptophan homeostasis.
Frontiers in molecular biosciencesScreening and identification of differential metabolites in serum and urine of bamaxiang pigs bitten by trimeresurus stejnegeri based on UPLC-Q-TOF/MS metabolomics technology.
The Journal of toxicological sciencesHartnup disease presenting as hereditary spastic paraplegia and severe peripheral neuropathy.
American journal of medical genetics. Part AInborn Errors of Metabolism Associated With Autism Spectrum Disorders: Approaches to Intervention.
Frontiers in neuroscienceBiochemical phenotyping of multiple myeloma patients at diagnosis reveals a disorder of mitochondrial complexes I and II and a Hartnup-like disturbance as underlying conditions, also influencing different stages of the disease.
Scientific reportsCOVID-19 and Hartnup disease: an affair of intestinal amino acid malabsorption.
Eating and weight disorders : EWDLoss of CLTRN function produces a neuropsychiatric disorder and a biochemical phenotype that mimics Hartnup disease.
American journal of medical genetics. Part AAssociation of Schizophrenia Risk With Disordered Niacin Metabolism in an Indian Genome-wide Association Study.
JAMA psychiatryIdentification and Characterization of Inhibitors of a Neutral Amino Acid Transporter, SLC6A19, Using Two Functional Cell-Based Assays.
SLAS discovery : advancing life sciences R & DStudy of Seizure-Manifested Hartnup Disorder Case Induced By Novel Mutations in SLC6A19.
Open life sciencesAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Hartnup.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Hartnup
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Dermatology-related disorders named after patients.
- Hartnup disease-causing SLC6A19 mutations lead to B0AT1 aberrant trafficking and ACE2 mis-localisation implicating the endoplasmic reticulum protein quality control.
- Dermatology-related disorders named after patients.
- Hartnup Disease-Associated Psychosis: A Dwindling Phenomenon or Just Underreported?
- Adult Neuropsychiatric Manifestation of Hartnup Disease With a Novel SLCA6A19 Variant: A Case Report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2116(Orphanet)
- OMIM OMIM:234500(OMIM)
- MONDO:0009324(MONDO)
- GARD:6569(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q200985(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Hartnup
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata