A deficiência do Complexo II é uma doença mitocondrial. As mitocôndrias são como pequenas fábricas dentro das nossas células que produzem mais de 90% da energia que o corpo precisa. Nas doenças mitocondriais, as mitocôndrias não funcionam direito, o que causa pouca energia na célula, danos às células e até a morte delas. Os sinais e sintomas da deficiência do complexo II podem variar muito, desde sintomas graves que colocam a vida em risco na infância até doenças musculares que começam na idade adulta. A deficiência do Complexo II pode ser causada por alterações nos genes SDHA, SDHB, SDHD ou SDHAF1. Em muitos casos, não é possível identificar as alterações genéticas que causam a doença. A deficiência do Complexo II é herdada de forma autossômica recessiva. Pessoas que possuem a alteração genética da deficiência do Complexo II podem ter um risco maior de desenvolver certos tipos de câncer.
Introdução
O que você precisa saber de cara
A deficiência do Complexo II é uma doença mitocondrial. As mitocôndrias são como pequenas fábricas dentro das nossas células que produzem mais de 90% da energia que o corpo precisa. Nas doenças mitocondriais, as mitocôndrias não funcionam direito, o que causa pouca energia na célula, danos às células e até a morte delas. Os sinais e sintomas da deficiência do complexo II podem variar muito, desde sintomas graves que colocam a vida em risco na infância até doenças musculares que começam na idade adulta. A deficiência do Complexo II pode ser causada por alterações nos genes SDHA, SDHB, SDHD ou SDHAF1. Em muitos casos, não é possível identificar as alterações genéticas que causam a doença. A deficiência do Complexo II é herdada de forma autossômica recessiva. Pessoas que possuem a alteração genética da deficiência do Complexo II podem ter um risco maior de desenvolver certos tipos de câncer.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 21 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 78 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisFlavoprotein (FP) subunit of succinate dehydrogenase (SDH) that is involved in complex II of the mitochondrial electron transport chain and is responsible for transferring electrons from succinate to ubiquinone (coenzyme Q) (PubMed:10746566, PubMed:24781757). SDH also oxidizes malate to the non-canonical enol form of oxaloacetate, enol-oxaloacetate (By similarity). Enol-oxaloacetate, which is a potent inhibitor of the succinate dehydrogenase activity, is further isomerized into keto-oxaloacetate
Mitochondrion inner membrane
Mitochondrial complex II deficiency, nuclear type 1
A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations. Clinical features include psychomotor regression in infants, poor growth with lack of speech development, severe spastic quadriplegia, dystonia, progressive leukoencephalopathy, muscle weakness, exercise intolerance, cardiomyopathy. Some patients manifest Leigh syndrome or Kearns-Sayre syndrome. MC2DN1 inheritance is autosomal recessive.
Plays an essential role in the assembly of succinate dehydrogenase (SDH), an enzyme complex (also referred to as respiratory complex II) that is a component of both the tricarboxylic acid (TCA) cycle and the mitochondrial electron transport chain, and which couples the oxidation of succinate to fumarate with the reduction of ubiquinone (coenzyme Q) to ubiquinol (PubMed:19465911, PubMed:24954417). Promotes maturation of the iron-sulfur protein subunit SDHB of the SDH catalytic dimer, protecting i
Mitochondrion matrix
Mitochondrial complex II deficiency, nuclear type 2
A form of mitochondrial complex II deficiency, a disorder with heterogeneous clinical manifestations. Some patients have multisystem involvement of the brain, heart, muscle, liver, and kidneys resulting in death in infancy, whereas others have only isolated cardiac or muscle involvement with onset in adulthood and normal cognition. Clinical features include psychomotor regression in infants, poor growth with lack of speech development, severe spastic quadriplegia, dystonia, progressive leukoencephalopathy, muscle weakness, exercise intolerance, cardiomyopathy. Some patients manifest Leigh syndrome or Kearns-Sayre syndrome. MC2DN2 inheritance is autosomal recessive.
Iron-sulfur protein (IP) subunit of the succinate dehydrogenase complex (mitochondrial respiratory chain complex II), responsible for transferring electrons from succinate to ubiquinone (coenzyme Q) (PubMed:26925370, PubMed:27604842). SDH also oxidizes malate to the non-canonical enol form of oxaloacetate, enol-oxaloacetate (By similarity). Enol-oxaloacetate, which is a potent inhibitor of the succinate dehydrogenase activity, is further isomerized into keto-oxaloacetate (By similarity)
Mitochondrion inner membrane
Pheochromocytoma/paraganglioma syndrome 4
A form of pheochromocytoma/paraganglioma syndrome, a tumor predisposition syndrome characterized by the development of neuroendocrine tumors, usually in adulthood. Pheochromocytomas are catecholamine-producing tumors that arise from chromaffin cells in the adrenal medulla. Paragangliomas develop from sympathetic paraganglia in the thorax, abdomen, and pelvis, as well as from parasympathetic paraganglia in the head and neck. PPGL4 inheritance is autosomal dominant.
Membrane-anchoring subunit of succinate dehydrogenase (SDH) that is involved in complex II of the mitochondrial electron transport chain and is responsible for transferring electrons from succinate to ubiquinone (coenzyme Q) (PubMed:10482792, PubMed:9533030). SDH also oxidizes malate to the non-canonical enol form of oxaloacetate, enol-oxaloacetate (By similarity). Enol-oxaloacetate, which is a potent inhibitor of the succinate dehydrogenase activity, is further isomerized into keto-oxaloacetate
Mitochondrion inner membrane
Pheochromocytoma/paraganglioma syndrome 1
A form of pheochromocytoma/paraganglioma syndrome, a tumor predisposition syndrome characterized by the development of neuroendocrine tumors, usually in adulthood. Pheochromocytomas are catecholamine-producing tumors that arise from chromaffin cells in the adrenal medulla. Paragangliomas develop from sympathetic paraganglia in the thorax, abdomen, and pelvis, as well as from parasympathetic paraganglia in the head and neck. PPGL1 inheritance is autosomal dominant.
Variantes genéticas (ClinVar)
1.211 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Deficiência de succinato-CoQ redutase isolada
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Mostrando amostra de 6 publicações de um total de 24
Two Patients Diagnosed as Succinate Dehydrogenase Deficiency: Case Report.
Succinate dehydrogenase deficiency, also known as mitochondrial complex II deficiency, is a rare inborn error of metabolism, accounting for approximately 2% of mitochondrial disease. Mutations in the four genes SDHA, B, C,and D have been reported resulting in diverse clinical presentations. The vast majority of clinically affected individuals reported in the literature harbor genetic variants within the SDHA gene and present with a Leigh syndrome phenotype, clinically defined as a subacute necrotizing encephalopathy. Herein, we report the first case of a 7-year-old child who was diagnosed as having succinate dehydrogenase deficiency. The affected child presented at 1 year of age with encephalopathy and developmental regression following viral illnesses. MRI changes supported a clinical diagnosis of Leigh syndrome and c.1328C>Q and c.872A>C SDHA variants were identified as compound heterozygous. Mitochondrial cocktail treatment including L-carnitine, riboflavin, thiamine, biotin, and ubiquinone was started. Mild clinical improvement was observed after treatment. He is now unable to walk and speak. The second patient, a 21-year-old woman, presented with generalized muscle weakness, easy fatigability, and cardiomyopathy. Investigations revealed increased lactate level of 67.4 mg/dL (4.5-19.8) with repeatedly increased plasma alanine levels 1,272 µmol/L (200-579). We administered carnitine, coenzyme, riboflavin, and thiamine for empirical therapy with the suspicion of mitochondrial disease. Clinical exome sequencing revealed compound heterozygous variants NM_004168.4:c.1945_1946del (p.Leu649GlufsTer4) at exon 15 of the SDHA gene and NM_004168.4:c.1909-12_1909-11del at intron 14 of SDHA gene. There are several very different presentations including Leigh syndrome, epileptic encephalopathy, and cardiomyopathy. Some cases present following viral illness; this feature is not specific to mitochondrial complex II deficiency and occurs in many other mitochondrial disease presentations. There is no cure for complex II deficiency, though some reported patients showed clinical improvement following riboflavin therapy. Riboflavin is not the only therapeutic intervention that is available to patients with an isolated complex II deficiency and various other compounds have shown promise in the treatment of symptoms, including L-carnitine and ubiquinone. Treatment alternatives such as parabenzoquinone EPI-743 and rapamycin are under study in the treatment of the disease.
SDHx mutation and pituitary adenoma: can in vivo 1H-MR spectroscopy unravel the link?
Germline mutations in genes encoding succinate dehydrogenase (SDH) are frequently involved in pheochromocytoma/paraganglioma (PPGL) development and were implicated in patients with the '3PAs' syndrome (associating pituitary adenoma (PA) and PPGL) or isolated PA. However, the causality link between SDHx mutation and PA remains difficult to establish, and in vivo tools for detecting hallmarks of SDH deficiency are scarce. Proton magnetic resonance spectroscopy (1H-MRS) can detect succinate in vivo as a biomarker of SDHx mutations in PGL. The objective of this study was to demonstrate the causality link between PA and SDH deficiency in vivo using 1H-MRS as a novel noninvasive tool for succinate detection in PA. Three SDHx-mutated patients suffering from a PPGL and a macroprolactinoma and one patient with an apparently sporadic non-functioning pituitary macroadenoma underwent MRI examination at 3 T. An optimized 1H-MRS semi-LASER sequence (TR = 2500 ms, TE = 144 ms) was employed for the detection of succinate in vivo. Succinate and choline-containing compounds were identified in the MR spectra as single resonances at 2.44 and 3.2 ppm, respectively. Choline compounds were detected in all the tumors (three PGL and four PAs), while a succinate peak was only observed in the three macroprolactinomas and the three PGL of SDHx-mutated patients, demonstrating SDH deficiency in these tumors. In conclusion, the detection of succinate by 1H-MRS as a hallmark of SDH deficiency in vivo is feasible in PA, laying the groundwork for a better understanding of the biological link between SDHx mutations and the development of these tumors.
A novel bi-allelic variant in the SDHB gene causes a severe mitochondrial complex II deficiency: a case report.
Isolated deficiency of complex II is a rare inborn error of metabolism, accounting for approximately 2% of mitochondrial diseases. Mitochondrial complex II deficiency is predominantly seen in cases with bi-allelic SDHA mutations. To our knowledge, only 11 patients and five pathogenic variants have been reported for the SDHB gene. Our patient had a severe clinical presentation with seizures and sepsis, and died at the age of 2 months. Muscle biopsy analysis was compatible with mitochondrial myopathy with complex II deficiency. The family was given a molecular diagnosis for their child 2 years after his death via a clinical exome test of a frozen muscle biopsy specimen and a novel homozygous missense variant c.592 A>G (p.Ser198Gly) in SDHB gene was detected by next-generation sequencing. Here, we present another patient with a novel homozygous SDHB variant causing severe complex II deficiency and early death.
Consolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency.
Isolated mitochondrial complex II deficiency is a rare cause of mitochondrial respiratory chain disease. To date biallelic variants in three genes encoding mitochondrial complex II molecular components have been unequivocally associated with mitochondrial disease (SDHA/SDHB/SDHAF1). Additionally, variants in one further complex II component (SDHD) have been identified as a candidate cause of isolated mitochondrial complex II deficiency in just two unrelated affected individuals with clinical features consistent with mitochondrial disease, including progressive encephalomyopathy and lethal infantile cardiomyopathy. We present clinical and genomic investigations in four individuals from an extended Palestinian family with clinical features consistent with an autosomal recessive mitochondrial complex II deficiency, in which our genomic studies identified a homozygous NM_003002.3:c.[205 G > A];[205 G > A];p.[(Glu69Lys)];[(Glu69Lys)] SDHD variant as the likely cause. Reviewing previously published cases, these findings consolidate disruption of SDHD function as a cause of mitochondrial complex II deficiency and further define the phenotypic spectrum associated with SDHD gene variants.
The genetic basis of isolated mitochondrial complex II deficiency.
Mitochondrial complex II (succinate:ubiquinone oxidoreductase) is the smallest complex of the oxidative phosphorylation system, a tetramer of just 140 kDa. Despite its diminutive size, it is a key complex in two coupled metabolic pathways - it oxidises succinate to fumarate in the tricarboxylic acid cycle and the electrons are used to reduce FAD to FADH2, ultimately reducing ubiquinone to ubiquinol in the respiratory chain. The biogenesis and assembly of complex II is facilitated by four ancillary proteins, all of which are autosomally-encoded. Numerous pathogenic defects have been reported which describe two broad clinical manifestations, either susceptibility to cancer in the case of single, heterozygous germline variants, or a mitochondrial disease presentation, almost exclusively due to bi-allelic recessive variants and associated with an isolated complex II deficiency. Here we present a compendium of pathogenic gene variants that have been documented in the literature in patients with an isolated mitochondrial complex II deficiency. To date, 61 patients are described, harbouring 32 different pathogenic variants in four distinct complex II genes: three structural subunit genes (SDHA, SDHB and SDHD) and one assembly factor gene (SDHAF1). Many pathogenic variants result in a null allele due to nonsense, frameshift or splicing defects however, the missense variants that do occur tend to induce substitutions at highly conserved residues in regions of the proteins that are critical for binding to other subunits or substrates. There is phenotypic heterogeneity associated with defects in each complex II gene, similar to other mitochondrial diseases.
Publicações recentes
Epilepsy Phenotype and EEG Finding of Rhythmic High-Amplitude Delta With Superimposed Spikes (RHADS) in Succinate Dehydrogenase Deficiency.
High Succinate peak in Magnetic Resonance Spectroscopy: A Diagnostic Clue for the Leukoencephalopathy Result from Succinate Dehydrogenase Deficiencies.
Two Patients Diagnosed as Succinate Dehydrogenase Deficiency: Case Report.
Cardiac disruption of SDHAF4-mediated mitochondrial complex II assembly promotes dilated cardiomyopathy.
Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation.
📚 EuropePMCmostrando 6
Two Patients Diagnosed as Succinate Dehydrogenase Deficiency: Case Report.
Molecular syndromologySDHx mutation and pituitary adenoma: can in vivo 1H-MR spectroscopy unravel the link?
Endocrine-related cancerA novel bi-allelic variant in the SDHB gene causes a severe mitochondrial complex II deficiency: a case report.
Clinical neurology and neurosurgeryConsolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency.
European journal of human genetics : EJHGThe genetic basis of isolated mitochondrial complex II deficiency.
Molecular genetics and metabolismA recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency.
Human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Deficiência de succinato-CoQ redutase isolada.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Deficiência de succinato-CoQ redutase isolada
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Two Patients Diagnosed as Succinate Dehydrogenase Deficiency: Case Report.
- SDHx mutation and pituitary adenoma: can in vivo 1H-MR spectroscopy unravel the link?
- A novel bi-allelic variant in the SDHB gene causes a severe mitochondrial complex II deficiency: a case report.
- Consolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency.
- The genetic basis of isolated mitochondrial complex II deficiency.
- Epilepsy Phenotype and EEG Finding of Rhythmic High-Amplitude Delta With Superimposed Spikes (RHADS) in Succinate Dehydrogenase Deficiency.
- High Succinate peak in Magnetic Resonance Spectroscopy: A Diagnostic Clue for the Leukoencephalopathy Result from Succinate Dehydrogenase Deficiencies.
- Cardiac disruption of SDHAF4-mediated mitochondrial complex II assembly promotes dilated cardiomyopathy.
- Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3208(Orphanet)
- OMIM OMIM:252011(OMIM)
- MONDO:0100294(MONDO)
- GARD:5053(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q23542368(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
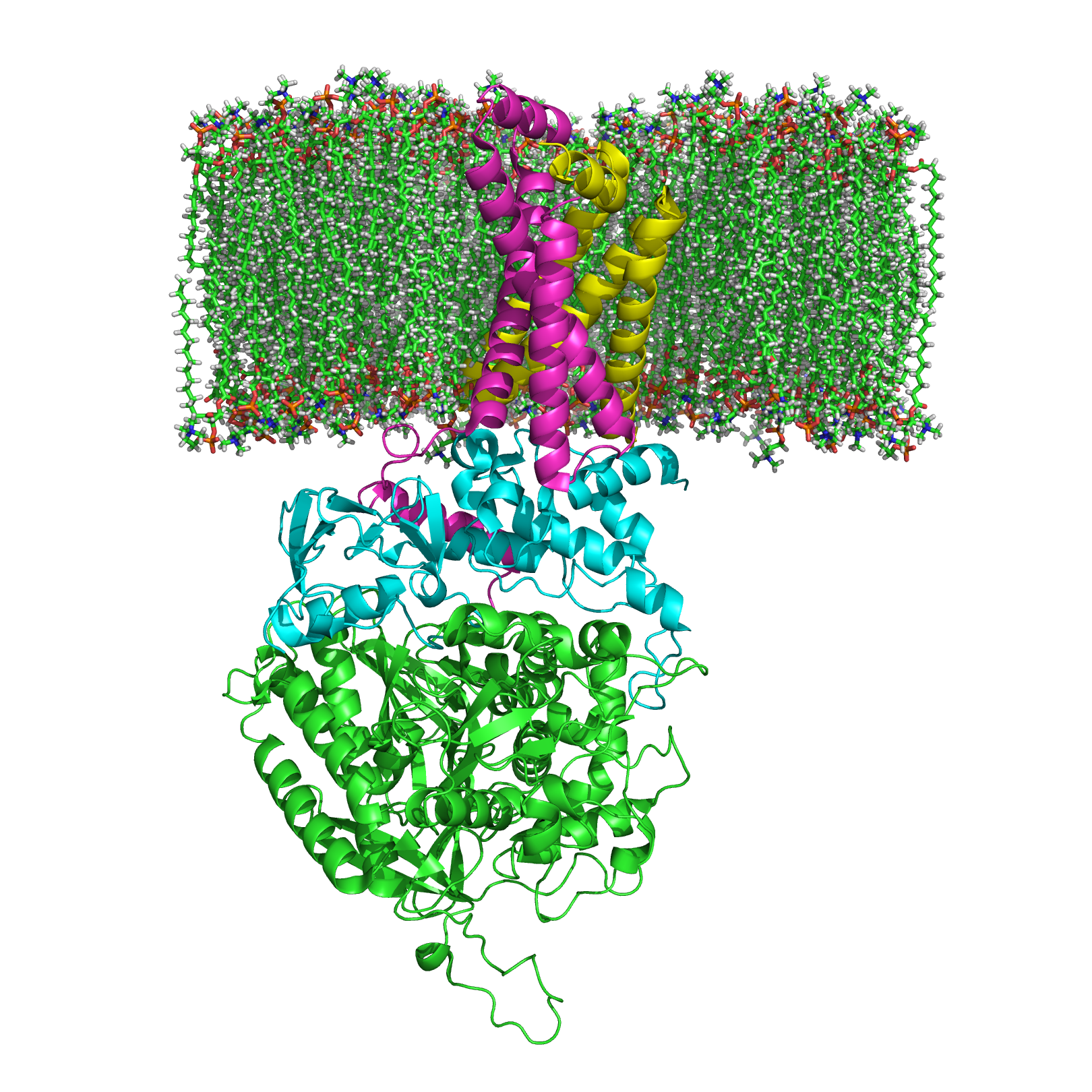
Deficiência de succinato-CoQ redutase isolada
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata