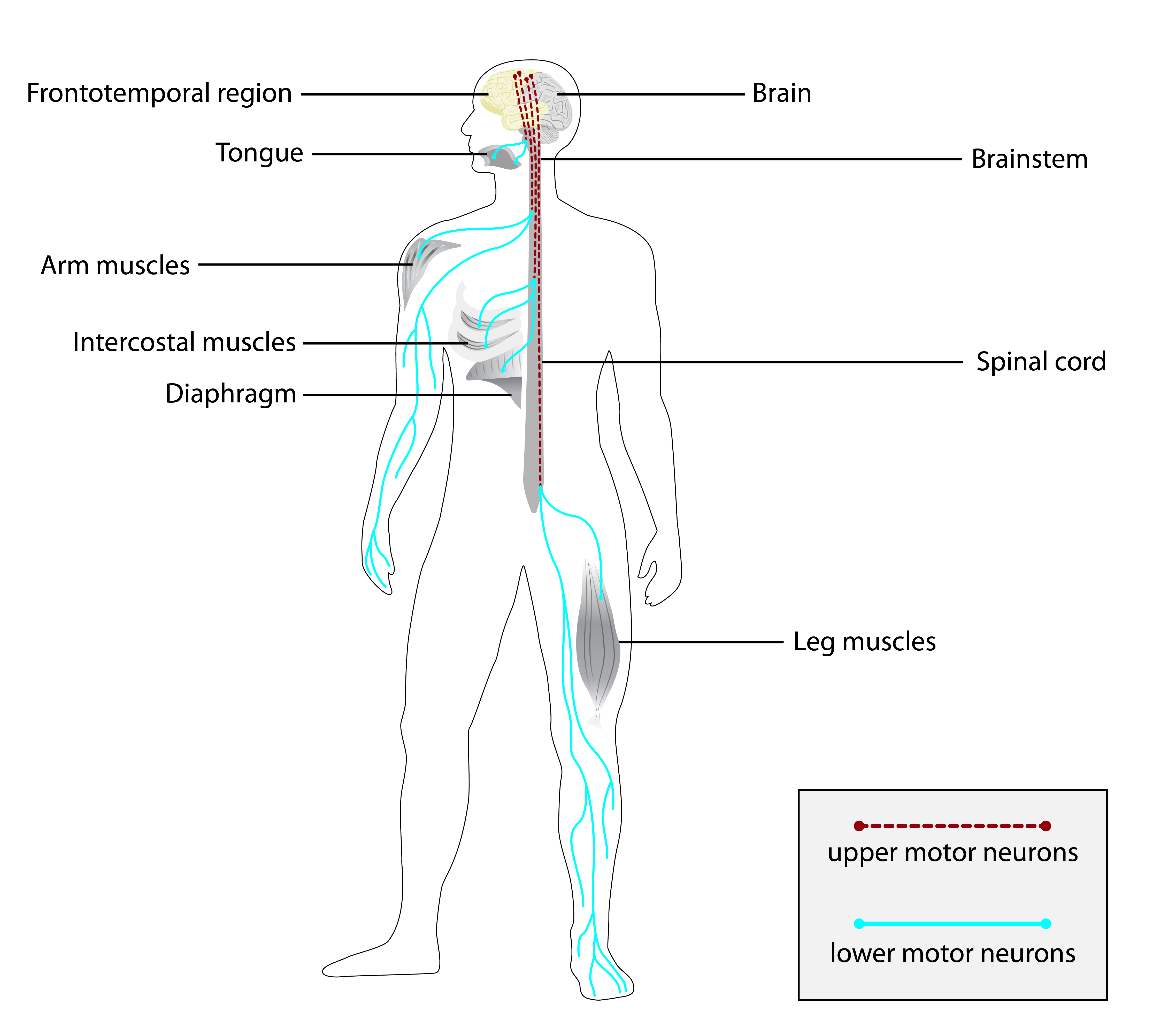
A esclerose lateral primária juvenil (JPLS) é uma doença do neurônio motor muito rara, caracterizada por disfunção progressiva do neurônio motor superior, levando à perda da capacidade de andar com dependência de cadeira de rodas e, subsequentemente, à perda da produção motora da fala.
Introdução
O que você precisa saber de cara
A esclerose lateral primária juvenil (JPLS) é uma doença do neurônio motor muito rara, caracterizada por disfunção progressiva do neurônio motor superior, levando à perda da capacidade de andar com dependência de cadeira de rodas e, subsequentemente, à perda da produção motora da fala.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 12 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 30 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisMay act as a GTPase regulator. Controls survival and growth of spinal motoneurons (By similarity)
Amyotrophic lateral sclerosis 2
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Component of the ERLIN1/ERLIN2 complex which mediates the endoplasmic reticulum-associated degradation (ERAD) of inositol 1,4,5-trisphosphate receptors (IP3Rs) such as ITPR1 (PubMed:17502376, PubMed:19240031). Promotes sterol-accelerated ERAD of HMGCR probably implicating an AMFR/gp78-containing ubiquitin ligase complex (PubMed:21343306). Involved in regulation of cellular cholesterol homeostasis by regulation the SREBP signaling pathway. May promote ER retention of the SCAP-SREBF complex (PubMe
Endoplasmic reticulum membrane
Spastic paraplegia 18B, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG18B is a severe form with onset in early childhood. Most affected individuals have severe psychomotor retardation. Some may develop significant joint contractures.
Variantes genéticas (ClinVar)
317 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 32 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
7 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Esclerose lateral primária juvenil
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Phenotype and Genotype of Children with ALS2 gene-Related Disorder.
The Alsin Rho Guanine Nucleotide Exchange Factor (ALS2) gene encodes a protein alsin that functions as a guanine nucleotide exchange factor. The variations in ALS2 gene leads to degeneration of upper motor neurons of the corticospinal tract. The phenotypes resulting from variants in ALS2 gene are infantile-onset ascending hereditary spastic paralysis (IAHSP, OMIM # 607225), juvenile primary lateral sclerosis (JPLS, OMIM # 606353), and juvenile amyotrophic lateral sclerosis (JALS, OMIM # 205100). Our study objectives were to describe the clinical phenotype and genotype of children with an established diagnosis of ALS2 gene-related disorder. The clinical details, laboratory data, and genotype findings of children with an established diagnosis of ALS2 gene-related disorder were collected from the hospital electronic database after obtaining institutional review board approval. One family with three affected siblings, a second family with a proband and an affected fetus, and a third family with two affected siblings with ALS2 gene variants were identified. IAHSP was diagnosed in all of our patients with variants in ALS2 gene. The clinical findings observed in our patients were insidious onset progressive spastic paraparesis, contractures, and dysarthria. Nonsense variants were observed in four patients while frameshift variant was observed in one family. Novel variants in ALS2 gene were identified in two unrelated families. ALS2 mutation results in rare neurodegenerative disorders with the clinical spectrum encompassing IAHSP, JPLS, and JALS disorders. In view of allelic heterogeneity described in the literature, more research studies are needed for establishing genotype-phenotype correlation in patients with ALS2 gene-related disorder.
Clinical and molecular spectrum of a large Egyptian cohort with ALS2-related disorders of infantile-onset of clinical continuum IAHSP/JPLS.
This study presents 46 patients from 23 unrelated Egyptian families with ALS2-related disorders without evidence of lower motor neuron involvement. Age at onset ranged from 10 months to 2.5 years, featuring progressive upper motor neuron signs. Detailed clinical phenotypes demonstrated inter- and intrafamilial variability. We identified 16 homozygous disease-causing ALS2 variants; sorted as splice-site, missense, frameshift, nonsense and in-frame in eight, seven, four, three, and one families, respectively. Seven of these variants were novel, expanding the mutational spectrum of the ALS2 gene. As expected, clinical severity was positively correlated with disease onset (p = 0.004). This work provides clinical and molecular profiles of a large single ethnic cohort of patients with ALS2 mutations, and suggests that infantile ascending hereditary spastic paralysis (IAHSP) and juvenile primary lateral sclerosis (JPLS) are belonged to one entity with no phenotype-genotype correlation.
ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules.
Infantile-onset Ascending Hereditary Spastic Paralysis, Juvenile Primary Lateral Sclerosis and Juvenile Amyotrophic Lateral Sclerosis are all motor neuron diseases related to mutations on the ALS2 gene, encoding for a 1657 amino acids protein named Alsin. This ~185 kDa multi-domain protein is ubiquitously expressed in various human tissues, mostly in the brain and the spinal cord. Several investigations have indicated how mutations within Alsin's structured domains may be responsible for the alteration of Alsin's native oligomerization state or Alsin's propensity to interact with protein partners. In this review paper, we propose a description of differences and similarities characterizing the above-mentioned ALS2-related rare neurodegenerative disorders, pointing attention to the effects of ALS2 mutation from molecule to organ and at the system level. Known cases were collected through a literature review and rationalized to deeply elucidate the neurodegenerative clinical outcomes as consequences of ALS2 mutations.
AI-based protein structure databases have the potential to accelerate rare diseases research: AlphaFoldDB and the case of IAHSP/Alsin.
Artificial intelligence (AI)-based protein structure databases are expected to have an impact on drug discovery. Here, we show how AlphaFold could support rare diseases research programs. We focus on Alsin, a protein responsible for rare motor neuron diseases, such as infantile-onset ascending hereditary spastic paralysis (IAHSP) and juvenile primary lateral sclerosis (JPLS), and involved in some cases of amyotrophic lateral sclerosis (ALS). First, we compared the AlphaFoldDB human Alsin model with homology models of Alsin domains. We then evaluated the flexibility profile of Alsin and of experimentally characterized mutants present in patients with IAHSP. Next, we compared preliminary models of dimeric/tetrameric Alsin responsible for its physiological action with hypothetical models reported in the literature. Finally, we suggest the best animal model for drug candidates testing. Overall, we computationally show that drug discovery efforts toward Alsin-involving diseases should be pursued.
The expanding clinical and genetic spectrum of alsin-related disorders: the first cohort of Brazilian patients.
There are three types of autosomal recessive disorders involving pathogenic variants in the ALS2 gene (OMIM*606352), infantile ascending hereditary spastic paraplegia (IAHSP), juvenile primary lateral sclerosis (JPLS) and juvenile amyotrophic lateral sclerosis (JALS), which are rare and related to retrograde degeneration of motor neurons. ALS2 pathogenic variants are distributed widely across the entire coding sequence and mostly result in a loss of protein function. Rarely, patients with JALS have been reported with lower motor neuron involvement. Here, we report the first Brazilian cohort (six patients) of JPLS with novel ALS2 pathogenic variants, and we propose an expanding clinical and genetic spectrum of alsin-related disorders. A review of the literature in PubMed from 2001 to September 2020 allowed us to identify 26 publications about the three different phenotypes caused by ALS2 variants (only case reports or families), encompassing 35 nonrelated families. We compiled data (sex, age, age at onset, first symptoms, atypical clinical features, molecular data, and clinical evolution (improvement or death)) from these studies and analyzed them in a general context on the basis of demographic features. ALS2-related disorder involves retrograde degeneration of the upper motor neurons of the pyramidal tracts and comprises a clinical continuum of the following three phenotypes: Infantile ascending hereditary spastic paraplegia (IAHSP), characterized by onset of spasticity with increased reflexes and sustained clonus of the lower limbs within the first two years of life, progressive weakness and spasticity of the upper limbs by age seven to eight years, and wheelchair dependence in the second decade with progression toward severe spastic tetraparesis and a pseudobulbar syndrome caused by progressive cranial nerve involvement. Juvenile primary lateral sclerosis (JPLS), characterized by upper motor neuron findings of pseudobulbar palsy and spastic quadriplegia without dementia or cerebellar, extrapyramidal, or sensory signs. Juvenile amyotrophic lateral sclerosis (JALS or ALS2), characterized by onset between ages three and 20 years. All affected individuals show a spastic pseudobulbar syndrome (spasticity of speech and swallowing) together with spastic paraplegia. Some individuals are bedridden by age 12 to 50 years. The diagnosis of ALS2-related disorder is established in a proband with suggestive findings and biallelic pathogenic variants in ALS2 identified on molecular genetic testing. Treatment of manifestations: Management by multidisciplinary specialists including neurology, orthopedics, physical therapy, occupational therapy, speech and language therapy, and feeding specialists (gastroenterology, nutrition) is recommended. Physical and occupational therapy promote mobility and independence; use of computer technologies and devices can facilitate writing and voice communication. Early detection and treatment of hip dislocation and/or spine deformities can prevent further complications. Surveillance: Routine reevaluation by the multidisciplinary care providers to monitor progression of existing findings and development of new findings. ALS2-related disorder is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an ALS2 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once both ALS2 pathogenic variants have been identified in an affected family member, carrier testing for at-risk family members, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Publicações recentes
Phenotype and Genotype of Children with ALS2 gene-Related Disorder.
Clinical and molecular spectrum of a large Egyptian cohort with ALS2-related disorders of infantile-onset of clinical continuum IAHSP/JPLS.
ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules.
AI-based protein structure databases have the potential to accelerate rare diseases research: AlphaFoldDB and the case of IAHSP/Alsin.
The expanding clinical and genetic spectrum of alsin-related disorders: the first cohort of Brazilian patients.
📚 EuropePMC1 artigos no totalmostrando 9
Phenotype and Genotype of Children with ALS2 gene-Related Disorder.
NeuropediatricsClinical and molecular spectrum of a large Egyptian cohort with ALS2-related disorders of infantile-onset of clinical continuum IAHSP/JPLS.
Clinical geneticsALS2-Related Motor Neuron Diseases: From Symptoms to Molecules.
BiologyAI-based protein structure databases have the potential to accelerate rare diseases research: AlphaFoldDB and the case of IAHSP/Alsin.
Drug discovery todayThe expanding clinical and genetic spectrum of alsin-related disorders: the first cohort of Brazilian patients.
Amyotrophic lateral sclerosis & frontotemporal degenerationGenotype-phenotype correlation in seven motor neuron disease families with novel ALS2 mutations.
American journal of medical genetics. Part AClinical presentation and natural history of infantile-onset ascending spastic paralysis from three families with an ALS2 founder variant.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyAbsence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms.
Human molecular geneticsIdentification of two novel ALS2 mutations in infantile-onset ascending hereditary spastic paraplegia.
Amyotrophic lateral sclerosis & frontotemporal degenerationAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Esclerose lateral primária juvenil.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Esclerose lateral primária juvenil
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Phenotype and Genotype of Children with ALS2 gene-Related Disorder.
- Clinical and molecular spectrum of a large Egyptian cohort with ALS2-related disorders of infantile-onset of clinical continuum IAHSP/JPLS.
- ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules.
- AI-based protein structure databases have the potential to accelerate rare diseases research: AlphaFoldDB and the case of IAHSP/Alsin.
- The expanding clinical and genetic spectrum of alsin-related disorders: the first cohort of Brazilian patients.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:247604(Orphanet)
- OMIM OMIM:606353(OMIM)
- MONDO:0011663(MONDO)
- GARD:4485(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q6318969(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Esclerose lateral primária juvenil
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata