Qualquer Esclerose Lateral Amiotrófica (ELA) em que a causa da doença é uma mutação no gene SETX.
Introdução
O que você precisa saber de cara
Qualquer Esclerose Lateral Amiotrófica (ELA) em que a causa da doença é uma mutação no gene SETX.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
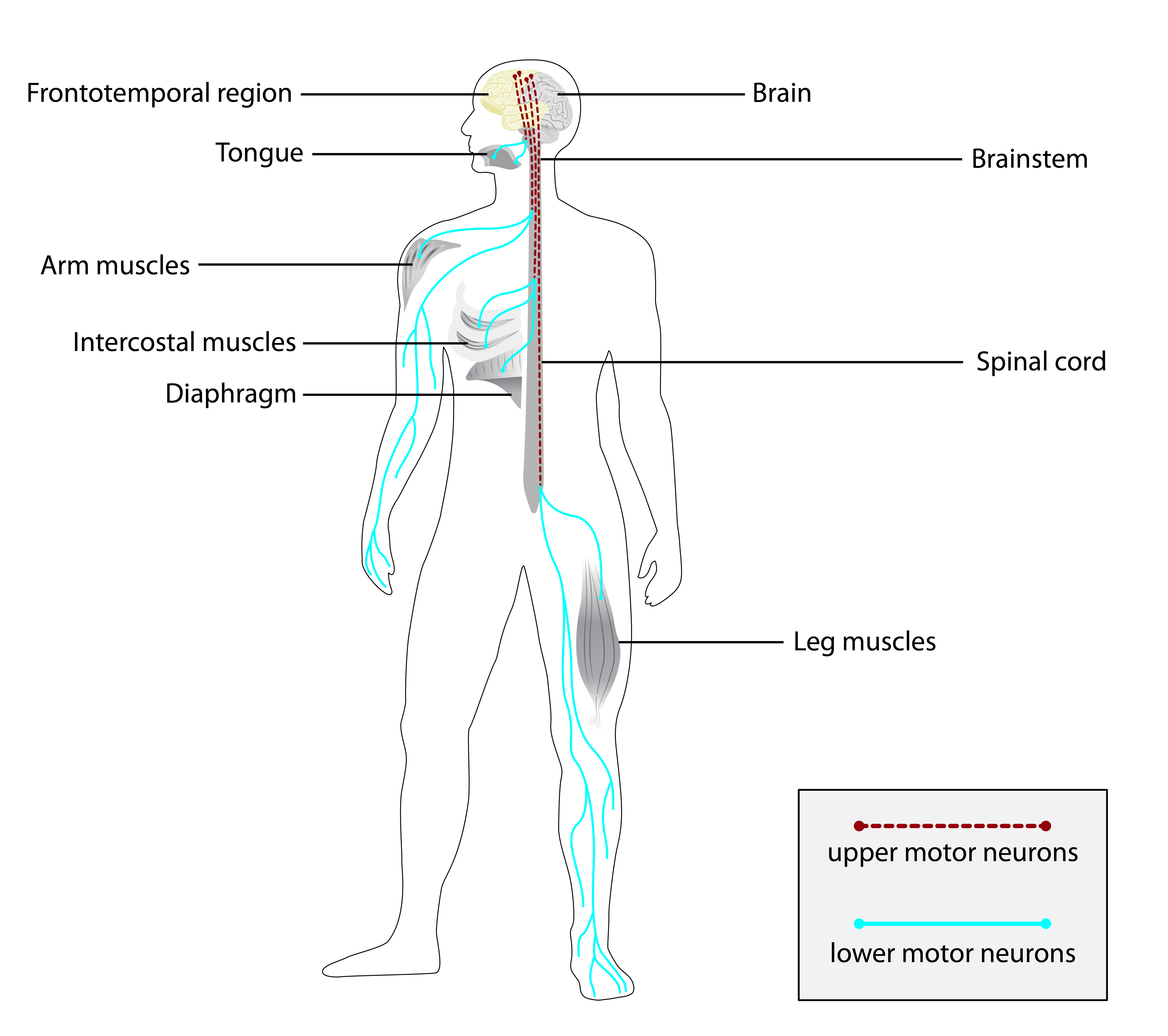
Partes do corpo afetadas
+ 20 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 26 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisATP-dependent 5'->3' DNA/RNA helicase that preferentially unwinds RNA substrates over DNA, playing a crucial role in resolving R-loops and promoting transcription termination (PubMed:36864660). Plays a role in transcription regulation by its ability to modulate RNA Polymerase II (Pol II) binding to chromatin and through its interaction with proteins involved in transcription (PubMed:19515850, PubMed:21700224). Contributes to the mRNA splicing efficiency and splice site selection (PubMed:19515850
NucleusNucleus, nucleoplasmNucleus, nucleolusCytoplasmChromosomeChromosome, telomereCell projection, axonCell projection, growth cone
Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 2
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN2 is an autosomal recessive form associated with peripheral neuropathy and elevated serum alpha-fetoprotein, immunoglobulins and, less commonly, creatine kinase levels. Some SCAN2 patients manifest oculomotor apraxia.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
375 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.502 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Esclerose lateral amiotrófica, tipo 4
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Allele-specific silencing of a dominant SETX mutation in familial amyotrophic lateral sclerosis type 4.
Amyotrophic lateral sclerosis 4 (ALS4) is an autosomal dominant motor neuron disease that is molecularly characterized by reduced R-loop levels and caused by pathogenic variants in senataxin (SETX). SETX encodes an RNA/DNA helicase that resolves three-stranded nucleic acid structures called R-loops. Currently, there are no disease-modifying therapies available for ALS4. Given that SETX is haplosufficient, removing the product of the mutated allele presents a potential therapeutic strategy. We designed a series of siRNAs to selectively target the RNA transcript from the ALS4 allele containing the c.1166T>C mutation (p.Leu389Ser). Transfection of HEK293 cells with siRNA and plasmids encoding either wild-type or mutant (Leu389Ser) epitope-tagged SETX revealed that three siRNAs specifically reduced mutant SETX protein levels while having minimal effect on the wild-type SETX protein. In ALS4 primary fibroblasts, siRNA treatment silenced the endogenous mutant SETX allele while sparing the wild-type allele and restored R-loop levels in patient cells. Our findings demonstrate that mutant SETX, differing from wild-type by a single nucleotide, can be effectively and specifically silenced by RNA interference.
Role of senataxin in R-loop-mediated neurodegeneration.
Senataxin is an RNA:DNA helicase that plays an important role in the resolution of RNA:DNA hybrids (R-loops) formed during transcription. R-loops are involved in the regulation of biological processes such as immunoglobulin class switching, gene expression and DNA repair. Excessive accumulation of R-loops results in DNA damage and loss of genomic integrity. Senataxin is critical for maintaining optimal levels of R-loops to prevent DNA damage and acts as a genome guardian. Within the nucleus, senataxin interacts with various RNA processing factors and DNA damage response and repair proteins. Senataxin interactors include survival motor neuron and zinc finger protein 1, with whom it co-localizes in sub-nuclear bodies. Despite its ubiquitous expression, mutations in senataxin specifically affect neurons and result in distinct neurodegenerative diseases such as amyotrophic lateral sclerosis type 4 and ataxia with oculomotor apraxia type 2, which are attributed to the gain-of-function and the loss-of-function mutations in senataxin, respectively. In addition, low levels of senataxin (loss-of-function) in spinal muscular atrophy result in the accumulation of R-loops causing DNA damage and motor neuron degeneration. Senataxin may play multiple functions in diverse cellular processes; however, its emerging role in R-loop resolution and maintenance of genomic integrity is gaining attention in the field of neurodegenerative diseases. In this review, we highlight the role of senataxin in R-loop resolution and its potential as a therapeutic target to treat neurodegenerative diseases.
Allele-specific silencing of a dominant SETX mutation in familial amyotrophic lateral sclerosis type 4.
Amyotrophic lateral sclerosis 4 (ALS4) is an autosomal dominant motor neuron disease that is molecularly characterized by reduced R-loop levels and caused by pathogenic variants in senataxin (SETX). SETX encodes an RNA/DNA helicase that resolves three-stranded nucleic acid structures called R-loops. Currently, there are no disease-modifying therapies available for ALS4. Given that SETX is haplosufficient, removing the product of the mutated allele presents a potential therapeutic strategy. We designed a series of siRNAs to selectively target the RNA transcript from the ALS4 allele containing the c.1166T>C mutation (p.Leu389Ser). Transfection of HEK293 cells with siRNA and plasmids encoding either wild-type or mutant (Leu389Ser) epitope tagged SETX revealed that three siRNAs specifically reduced mutant SETX protein levels without affecting the wild-type SETX protein. In ALS4 primary fibroblasts, siRNA treatment silenced the endogenous mutant SETX allele, while sparing the wild-type allele, and restored R-loop levels in patient cells. Our findings demonstrate that mutant SETX, differing from wild-type by a single nucleotide, can be effectively and specifically silenced by RNA interference, highlighting the potential of allele-specific siRNA as a therapeutic approach for ALS4.
Altered SYNJ2BP-mediated mitochondrial-ER contacts in motor neuron disease.
Synaptojanin 2 binding protein (SYNJ2BP) is an outer mitochondrial membrane protein with a cytosolic PDZ domain that functions as a cellular signaling hub. Few studies have evaluated its role in disease. Here we use induced pluripotent stem cell (iPSC)-derived motor neurons and post-mortem tissue from patients with two hereditary motor neuron diseases, spinal and bulbar muscular atrophy (SBMA) and amyotrophic lateral sclerosis type 4 (ALS4), and show that SYNJ2BP expression is increased in diseased motor neurons. Similarly, we show that SYNJ2BP expression increases in iPSC-derived motor neurons undergoing stress. Using proteomic analysis, we found that elevated SYNJ2BP alters the cellular distribution of mitochondria and increases mitochondrial-ER membrane contact sites. Furthermore, decreasing SYNJ2BP levels improves mitochondrial oxidative function in the diseased motor neurons. Together, our observations offer new insight into the molecular pathology of motor neuron disease and the role of SYNJ2BP in mitochondrial dysfunction.
Unusual electrophysiological findings in a Chinese ALS 4 family with SETX-L389S mutation: a three-year follow-up.
Amyotrophic lateral sclerosis type 4 (ALS4) is a familial form of ALS caused by mutations in the SETX gene. To date, there are seven unrelated ALS4 families with four missense mutations (L389S, T31I, R2136H, and M386T) in SETX. ALS4 is characterized by early onset, distal muscle weakness and atrophy, pyramidal signs, and the absence of sensory deficits. Motor conduction studies often present normality or reduced amplitudes of compound muscle action potential (CMAP). The conduction blocks (CBs) are rare and only observed in one male of an Italian ALS4 family. Our study showed that seven symptomatic patients presented the classical ALS4 phenotype with two asymptomatic females in a Chinese family spanning three generations. Sequencing analysis revealed a heterozygous c.1166T > C/p.L389S mutation in SETX that co-segregated with disease phenotype in the family. The same mutation has been identified previously in three ALS4 families from the United States and Italy, respectively. Specifically, three young males presented multiple CBs and abnormal temporal dispersions (TD) in the median, ulnar and tibial nerves over the three-year follow-up period. Moreover, for the first time, we found that senataxin was also expressed in the myelin sheath of peripheral nerves besides axons. The study indicates that CBs and abnormal TD are the characteristics in the ALS4 family, providing pivotal familial evidence of CBs and TD of motor nerves in ALS4. The unusual electrophysiological features may be associated with the expression of senataxin in peripheral nerves.
Publicações recentes
Allele-specific silencing of a dominant SETX mutation in familial amyotrophic lateral sclerosis type 4.
Allele-specific silencing of a dominant SETX mutation in familial amyotrophic lateral sclerosis type 4.
Role of senataxin in R-loop-mediated neurodegeneration.
Altered SYNJ2BP-mediated mitochondrial-ER contacts in motor neuron disease.
De novo pathogenic variant in SETX causes a rapidly progressive neurodegenerative disorder of early childhood-onset with severe axonal polyneuropathy.
📚 EuropePMC17.515 artigos no totalmostrando 12
Allele-specific silencing of a dominant SETX mutation in familial amyotrophic lateral sclerosis type 4.
HGG advancesRole of senataxin in R-loop-mediated neurodegeneration.
Brain communicationsAltered SYNJ2BP-mediated mitochondrial-ER contacts in motor neuron disease.
Neurobiology of diseaseDe novo pathogenic variant in SETX causes a rapidly progressive neurodegenerative disorder of early childhood-onset with severe axonal polyneuropathy.
Acta neuropathologica communicationsSUMOylated Senataxin functions in genome stability, RNA degradation, and stress granule disassembly, and is linked with inherited ataxia and motor neuron disease.
Molecular genetics & genomic medicineUnusual electrophysiological findings in a Chinese ALS 4 family with SETX-L389S mutation: a three-year follow-up.
Journal of neurologySETX (senataxin), the helicase mutated in AOA2 and ALS4, functions in autophagy regulation.
AutophagyTight expression regulation of senataxin, linked to motor neuron disease and ataxia, is required to avert cell-cycle block and nucleolus disassembly.
Heliyon[A case of motor and sensory polyneuropathy and respiratory failure with novel heterozygous mutation of the senataxin gene].
Rinsho shinkeigaku = Clinical neurologySenataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients.
Acta neuropathologicaSenataxin: Genome Guardian at the Interface of Transcription and Neurodegeneration.
Journal of molecular biologyHuman Senataxin Modulates Structural Plasticity of the Neuromuscular Junction in Drosophila through a Neuronally Conserved TGFβ Signalling Pathway.
Neuro-degenerative diseasesAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Esclerose lateral amiotrófica, tipo 4.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Esclerose lateral amiotrófica, tipo 4
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Allele-specific silencing of a dominant SETX mutation in familial amyotrophic lateral sclerosis type 4.
- Role of senataxin in R-loop-mediated neurodegeneration.
- Allele-specific silencing of a dominant SETX mutation in familial amyotrophic lateral sclerosis type 4.
- Altered SYNJ2BP-mediated mitochondrial-ER contacts in motor neuron disease.
- Unusual electrophysiological findings in a Chinese ALS 4 family with SETX-L389S mutation: a three-year follow-up.
- De novo pathogenic variant in SETX causes a rapidly progressive neurodegenerative disorder of early childhood-onset with severe axonal polyneuropathy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:357043(Orphanet)
- OMIM OMIM:602433(OMIM)
- MONDO:0011223(MONDO)
- GARD:10502(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18553715(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Esclerose lateral amiotrófica, tipo 4
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Ensaios clínicos
- fonte: ClinicalTrials.gov