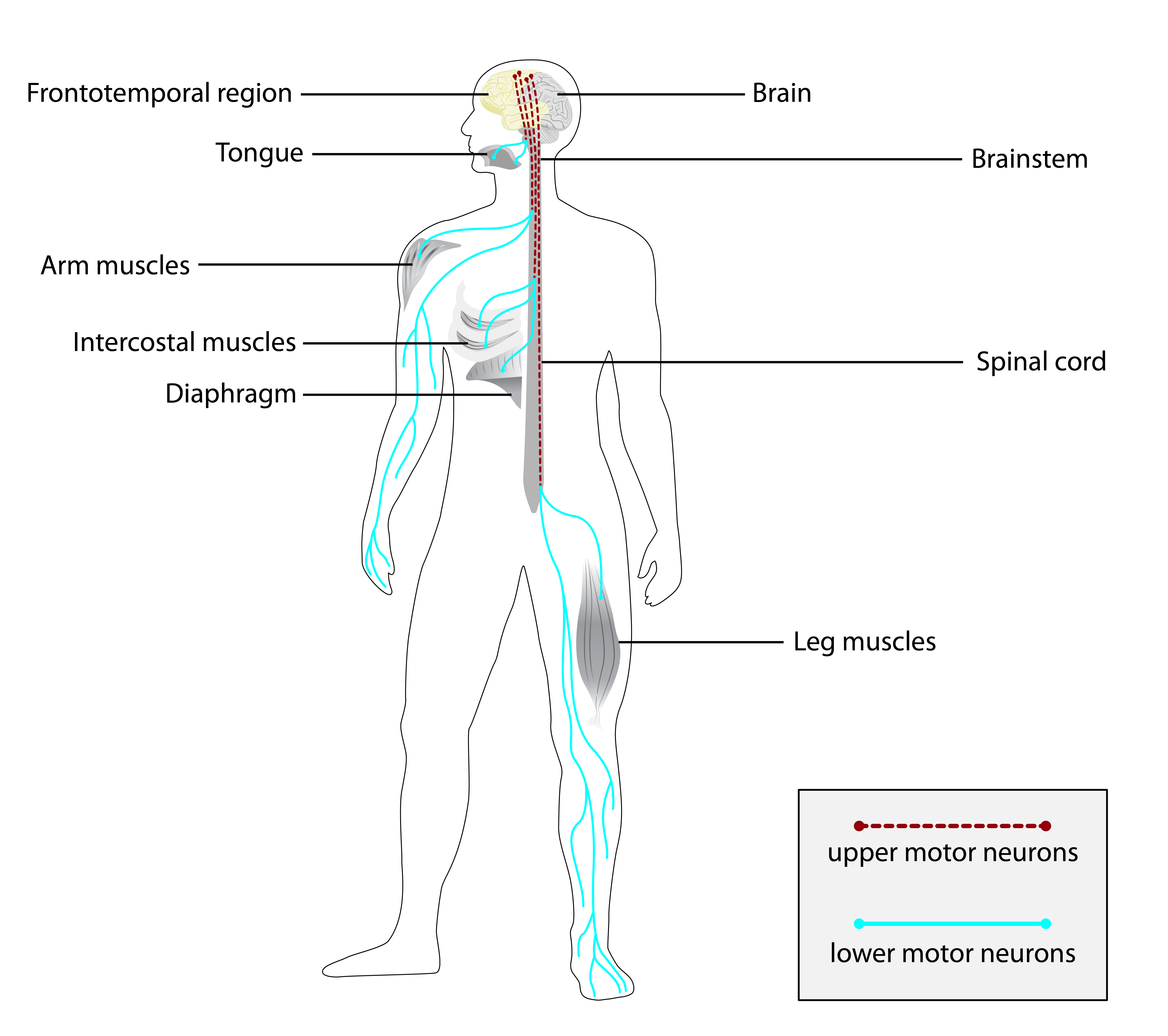
A esclerose lateral amiotrófica (ELA), também conhecida como doença do neurônio motor (DNM) ou doença de Lou Gehrig, é uma doença neurodegenerativa rara e terminal, definida pela perda progressiva de neurônios motores superiores e inferiores que normalmente controlam a contração muscular voluntária. A ELA é a mais comum das doenças do neurônio motor. A ELA frequentemente se manifesta em seus estágios iniciais com rigidez muscular gradual, espasmos, fraqueza e atrofia. A perda de neurônios motores continua tipicamente até que a capacidade de comer, falar, mover-se e respirar sem suporte mecânico seja perdida. Estima-se que pelo menos 50% das pessoas com ELA apresentem mudanças significativas no pensamento e no comportamento, sendo que 15% dos indivíduos desenvolvem demência frontotemporal.
Introdução
O que você precisa saber de cara
Visão geral
A síndrome semelhante à doença de Huntington por expansões C9ORF72 é uma doença neurodegenerativa rara, de herança autossômica dominante, que se manifesta na idade adulta. A condição recebe esse nome porque seus sintomas se assemelham aos da doença de Huntington, mas é causada por mutações em um gene diferente, o C9ORF72. A prevalência estimada é de menos de 1 caso por 1.000.000 de pessoas.[1][3]
Sinais e sintomas
Os sintomas mais comuns incluem coreia (movimentos involuntários e irregulares), parkinsonismo (rigidez, tremor, lentidão de movimentos), distonia (contrações musculares sustentadas), mioclonias (espasmos musculares rápidos), ataxia (falta de coordenação), disfunção do neurônio motor superior (fraqueza, espasticidade) e comprometimento cognitivo e de memória. Também podem ocorrer alterações psiquiátricas como depressão, ansiedade, psicose e comportamento inapropriado.[1][3]
Causas genéticas
A doença é causada por expansões no gene C9ORF72, localizado no cromossomo 9. Esse gene fornece instruções para a produção da proteína C9orf72, que atua como um fator de troca de nucleotídeos guanina (Guanine nucleotide exchange factor). A herança é autossômica dominante, o que significa que uma cópia alterada do gene é suficiente para causar a doença. A idade de início é na vida adulta.[1][4]
Diagnóstico
O diagnóstico é confirmado por meio de teste genético molecular, que identifica a expansão patogênica no gene C9ORF72. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis para esse fim. Atualmente, há 80 variantes associadas à doença registradas no ClinVar. O código CID-10 correspondente é G10, e a doença é classificada no MONDO como MONDO:0018425.[1][4]
Tratamento e manejo
Não há cura para a síndrome semelhante à doença de Huntington por expansões C9ORF72. O manejo é multidisciplinar e foca no alívio dos sintomas e na melhora da qualidade de vida. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para a doença, incluindo atendimento em reabilitação para doenças raras. O tratamento deve ser individualizado, com acompanhamento de neurologista, psiquiatra, fisioterapeuta e outros profissionais conforme a necessidade. Não há medicamentos específicos aprovados para esta condição.[1]
Prognóstico e qualidade de vida
O prognóstico é variável, mas a doença é progressiva e pode levar a comprometimento funcional significativo. O acompanhamento regular com equipe multidisciplinar e o suporte de reabilitação são fundamentais para manter a qualidade de vida pelo maior tempo possível.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A esclerose lateral amiotrófica (ELA), também conhecida como doença do neurônio motor (DNM) ou doença de Lou Gehrig, é uma doença neurodegenerativa rara e terminal, definida pela perda progressiva de neurônios motores superiores e inferiores que normalmente controlam a contração muscular voluntária. A ELA é a mais comum das doenças do neurônio motor. A ELA frequentemente se manifesta em seus estágios iniciais com rigidez muscular gradual, espasmos, fraqueza e atrofia. A perda de neurônios motores continua tipicamente até que a capacidade de comer, falar, mover-se e respirar sem suporte mecânico seja perdida. Estima-se que pelo menos 50% das pessoas com ELA apresentem mudanças significativas no pensamento e no comportamento, sendo que 15% dos indivíduos desenvolvem demência frontotemporal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A síndrome semelhante à doença de Huntington por expansões C9ORF72 é uma doença neurodegenerativa rara, de herança autossômica dominante, que se manifesta na idade adulta. A condição recebe esse nome porque seus sintomas se assemelham aos da doença de Huntington, mas é causada por mutações em um gene diferente, o C9ORF72. A prevalência estimada é de menos de 1 caso por 1.000.000 de pessoas.[1][3]
Sinais e sintomas
Os sintomas mais comuns incluem coreia (movimentos involuntários e irregulares), parkinsonismo (rigidez, tremor, lentidão de movimentos), distonia (contrações musculares sustentadas), mioclonias (espasmos musculares rápidos), ataxia (falta de coordenação), disfunção do neurônio motor superior (fraqueza, espasticidade) e comprometimento cognitivo e de memória. Também podem ocorrer alterações psiquiátricas como depressão, ansiedade, psicose e comportamento inapropriado.[1][3]
Causas genéticas
A doença é causada por expansões no gene C9ORF72, localizado no cromossomo 9. Esse gene fornece instruções para a produção da proteína C9orf72, que atua como um fator de troca de nucleotídeos guanina (Guanine nucleotide exchange factor). A herança é autossômica dominante, o que significa que uma cópia alterada do gene é suficiente para causar a doença. A idade de início é na vida adulta.[1][4]
Diagnóstico
O diagnóstico é confirmado por meio de teste genético molecular, que identifica a expansão patogênica no gene C9ORF72. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis para esse fim. Atualmente, há 80 variantes associadas à doença registradas no ClinVar. O código CID-10 correspondente é G10, e a doença é classificada no MONDO como MONDO:0018425.[1][4]
Tratamento e manejo
Não há cura para a síndrome semelhante à doença de Huntington por expansões C9ORF72. O manejo é multidisciplinar e foca no alívio dos sintomas e na melhora da qualidade de vida. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para a doença, incluindo atendimento em reabilitação para doenças raras. O tratamento deve ser individualizado, com acompanhamento de neurologista, psiquiatra, fisioterapeuta e outros profissionais conforme a necessidade. Não há medicamentos específicos aprovados para esta condição.[1]
Prognóstico e qualidade de vida
O prognóstico é variável, mas a doença é progressiva e pode levar a comprometimento funcional significativo. O acompanhamento regular com equipe multidisciplinar e o suporte de reabilitação são fundamentais para manter a qualidade de vida pelo maior tempo possível.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 4 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 14 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Acts as a guanine-nucleotide releasing factor (GEF) for Rab GTPases by promoting the conversion of inactive RAB-GDP to the active form RAB-GTP (PubMed:27103069, PubMed:27193190, PubMed:27617292, PubMed:28195531, PubMed:37821429). Acts as a GEF for RAB39A which enables HOPS-mediated autophagosome-lysosome membrane tethering and fusion in mammalian autophagy (PubMed:37821429). Component of the C9orf72-SMCR8 complex where both subunits display GEF activity and that regulates autophagy (PubMed:27103
CytoplasmNucleusCytoplasm, P-bodyCytoplasm, Stress granuleEndosomeLysosomeCytoplasmic vesicle, autophagosomeAutolysosomeSecretedCell projection, axonCell projection, growth conePerikaryonCell projection, dendritePresynapsePostsynapseNucleus membrane
Frontotemporal dementia and/or amyotrophic lateral sclerosis 1
An autosomal dominant neurodegenerative disorder characterized by adult onset of frontotemporal dementia and/or amyotrophic lateral sclerosis in an affected individual. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis.
Variantes genéticas (ClinVar)
80 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome semelhante à doença de Huntington por expansões C9ORF72
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Screening for the C9ORF72 expansion in Greek Huntington Disease phenocopies and controls and meta-analysis of current data.
Several European studies examined the role of C9orf72 repeat expansion in patients with Huntington-disease like phenotypes (HD-L). The scope of our study is to investigate the expansion frequency in a Greek HD-L cohort and the meta-analysis of all published cases. This will be of use in genetic counseling of these cases. A cohort of 74 patients with HD-L and 67 healthy controls were screened for the C9orf72 expansion status. Case-controls comparison was assessed with the Pearson's chi-square statistic for a 2 × 2 table.A systematic database search was conducted and seven studies, including the current study, were considered eligible for inclusion in a meta-analysis considering a total of 812 patients with HD phenocopies. Pooled mutation frequency was calculated using a Random Effects model or the Mantel-Haezsel fixed effects model, depending on the observed heterogeneity. In our cohort, one patient was found to have a pathologic expansion of C9orf72, and none from the control group (chi-square: 0.91, p-value: 0.34). Pooled mutation frequency was found at 2% (CI: 1-3%) with low heterogeneity (I2:15%). Based on this meta-analysis the recommendation for genetic testing for C9orf72 expansions is further solidified.
Investigations of Huntington's Disease and Huntington's Disease-Like Syndromes in Indian Choreatic Patients.
The diagnostic workup for choreiform movement disorders including Huntington's disease (HD) and those mimicking HD like phenotype is complex. The aim of the present study was to genetically define HD and HD-like presentations in an Indian cohort. We also describe HTT-CAG expansion manifesting as neuroferritinopathy-like disorder in four families from Punjab in India. 159 patients clinically diagnosed as HD and HD-like presentations from various tertiary neurology clinics were referred to our centre (CSIR-IGIB) for genetic investigations. As a first tier test, CAG-TNR for HTT was performed and subsequently HD-negative samples were screened for JPH3 (HDL2), TBP (SCA17), ATN1 (DRPLA), PPP2R2B (SCA12) and GGGGCC expansion in C9orf72 gene. Four families presenting as neuroferritinopathy-like disorder were also investigated for HTT-CAG expansion. 94 of 159 (59%) patients were found to have expanded HTT-CAG repeats. Pathogenic repeat expansion in JPH3, TBP, ATN1 and C9orf72 were not found in HD negative cases. Two patients were positive for SCA12-CAG expansion in pathogenic length, whereas 5 cases harboured TBP-CAG repeats falling in reduced penetrance range of 41- 48 repeats for SCA17. Four unrelated families, presented with atypical chorea and brain MRI findings suggestive of basal ganglia abnormalities mimicking neuroferritinopathy were found to harbour HTT-CAG expansion. We present SCA12 as a new reported phenocopy of HD which should be considered for diagnostic workout along with SCA17 for HD-like syndromes. This study also illustrates the necessity, to consider evolving HD like phenotype, as a clinical diagnosis for cases with initial manifestations depicting neuroferritinopathy.
A study of Huntington disease-like syndromes in black South African patients reveals a single SCA2 mutation and a unique distribution of normal alleles across five repeat loci.
Huntington disease (HD) is a progressive neurodegenerative disease, characterised by a triad of movement disorder, emotional and behavioural disturbances and cognitive impairment. The underlying cause is an expanded CAG repeat in the huntingtin gene. For a small proportion of patients presenting with HD-like symptoms, the mutation in this gene is not identified and they are said to have a HD "phenocopy". South Africa has the highest number of recorded cases of an African-specific phenocopy, Huntington disease-like 2 (HDL2), caused by a repeat expansion in the junctophilin-3 gene. However, a significant proportion of black patients with clinical symptoms suggestive of HD still test negative for HD and HDL2. This study thus aimed to investigate five other loci associated with HD phenocopy syndromes - ATN1, ATXN2, ATXN7, TBP and C9orf72. In a sample of patients in whom HD and HDL2 had been excluded, a single expansion was identified in the ATXN2 gene, confirming a diagnosis of Spinocerebellar ataxia 2. The results indicate that common repeat expansion disorders do not contribute significantly to the HD-like phenotype in black South African patients. Importantly, allele sizing reveals unique distributions of normal repeat lengths across the associated loci in the African population studied.
Publicações recentes
Screening for the C9ORF72 expansion in Greek Huntington Disease phenocopies and controls and meta-analysis of current data.
Investigations of Huntington's Disease and Huntington's Disease-Like Syndromes in Indian Choreatic Patients.
🥉 Relato de casoA study of Huntington disease-like syndromes in black South African patients reveals a single SCA2 mutation and a unique distribution of normal alleles across five repeat loci.
C9ORF72 mutations in neurodegenerative diseases.
Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population.
📚 EuropePMCmostrando 3
Screening for the C9ORF72 expansion in Greek Huntington Disease phenocopies and controls and meta-analysis of current data.
Tremor and other hyperkinetic movements (New York, N.Y.)Investigations of Huntington's Disease and Huntington's Disease-Like Syndromes in Indian Choreatic Patients.
Journal of Huntington's diseaseA study of Huntington disease-like syndromes in black South African patients reveals a single SCA2 mutation and a unique distribution of normal alleles across five repeat loci.
Journal of the neurological sciencesAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome semelhante à doença de Huntington por expansões C9ORF72.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome semelhante à doença de Huntington por expansões C9ORF72
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Screening for the C9ORF72 expansion in Greek Huntington Disease phenocopies and controls and meta-analysis of current data.
- Investigations of Huntington's Disease and Huntington's Disease-Like Syndromes in Indian Choreatic Patients.
- A study of Huntington disease-like syndromes in black South African patients reveals a single SCA2 mutation and a unique distribution of normal alleles across five repeat loci.
- C9ORF72 mutations in neurodegenerative diseases.
- Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:401901(Orphanet)
- MONDO:0018425(MONDO)
- GARD:21702(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55346046(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome semelhante à doença de Huntington por expansões C9ORF72
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata