Qualquer craniossinostose causada por uma mutação no gene ZIC1.
Introdução
O que você precisa saber de cara
Qualquer craniossinostose causada por uma mutação no gene ZIC1.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 13 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 26 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Curadoria gene-doença
fontes oficiaisActs as a transcriptional activator. Involved in neurogenesis. Plays important roles in the early stage of organogenesis of the CNS, as well as during dorsal spinal cord development and maturation of the cerebellum. Involved in the spatial distribution of mossy fiber (MF) neurons within the pontine gray nucleus (PGN). Plays a role in the regulation of MF axon pathway choice. Promotes MF migration towards ipsilaterally-located cerebellar territories. May have a role in shear flow mechanotransduct
NucleusCytoplasm
Craniosynostosis 6
A form of craniosynostosis, a primary abnormality of skull growth involving premature fusion of one or more cranial sutures. The growth velocity of the skull often cannot match that of the developing brain resulting in an abnormal head shape and, in some cases, increased intracranial pressure, which must be treated promptly to avoid permanent neurodevelopmental disability.
Variantes genéticas (ClinVar)
61 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de craniossinostose-anomalias esqueléticas e cerebelares-dificuldades de aprendizagem
Centros de Referência SUS
24 centros habilitados pelo SUS para Síndrome de craniossinostose-anomalias esqueléticas e cerebelares-dificuldades de aprendizagem
Centros para Síndrome de craniossinostose-anomalias esqueléticas e cerebelares-dificuldades de aprendizagem
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Znhit3 regulates p53/p21 signaling and governs cerebellar granule cell development.
Mutations in ZNHIT3 are strongly associated with progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO syndrome), characterized by severe cerebellar atrophy and profound intellectual disability; however, their role in cerebellar development remains unknown. By developing spatiotemporally-regulated conditional Znhit3 knockout mice, we discovered that Znhit3 is essential for granule cell progenitor survival, proliferation, differentiation, and migration. Knockout of Znhit3 caused loss of granule cell progenitors due to apoptosis, premature cell-cycle exit, and migration arrest and resulted in progressive anterior-lobe atrophy and motor deficits. The granule cell progenitor-autonomous defects secondarily impaired Purkinje cell alignment, dendritic maturation, and synaptic organization. Transcriptomic analyses revealed activation of the p53/p21 pathway, rRNA processing defects, and nucleolar stress. Genetic or pharmacologic inhibition of p53/p21 signaling rescued granule cell progenitor development and restored cerebellar architecture in the Znhit3-knockout mice. Thus, ZNHIT3 is a critical regulator of ribosome biogenesis and cerebellar growth, suggesting nucleolar stress-p53/p21 signaling as a potential therapeutic target in ZNHIT3-related disorders.
Child Neurology: Multiple Genetic Etiologies Causing Dandy-Walker Variant With Microcephaly, Epilepsy, and Global Developmental Delay.
Dandy-Walker syndrome is typically characterized by near-complete cerebellar vermis agenesis, enlarged posterior fossa, and dilated fourth ventricle. By contrast, Dandy-Walker variant (DWv) shows milder features, typically characterized by partial agenesis of the cerebellar vermis, mild enlargement of the posterior fossa, and variable dilation of the fourth ventricle. Both conditions are usually associated with normal or enlarged head circumference. We report a 16-month-old girl presenting with congenital microcephaly, frequent seizures, and severe global developmental delay. Brain MRI revealed findings consistent with DWv, which did not explain the severity of her clinical symptoms or her microcephaly. Chromosomal microarray analysis revealed multiple regions of homozygosity on chromosome 11, indicating potential recessive inheritance; karyotype analysis and mitochondrial testing showed no clear etiology. Trio-based whole-exome sequencing identified a heterozygous variant (NM_021096.4:c.4891T>A/p.Phe1631Ile) in CACNA1I and a homozygous variant (NM_002335.4:c.1310C>T/p.Thr437Met) in LRP5. Variants in CACNA1I are associated with neurodevelopmental disorders, including epilepsy and developmental delay, while variants in LRP5 are linked to osteoporosis and microcephaly. Based on the clinical presentation and molecular findings, we hypothesize that both variants contributed to the patient's complex phenotype. This case highlights that in patients with unusually severe or atypical manifestations, the possibility of multiple genetic pathogenic contributions should be considered, and comprehensive genomic evaluation is essential for accurate diagnosis and management.
PRPS1 (p.V42L) Mutation in Arts Syndrome Induces Aberrant Neural Stem Cell Development and Neuronal Senescence-Like Phenotype: Rescue by Nicotinamide Mononucleotide Supplementation.
Arts syndrome is a rare X-linked recessive neurodevelopmental disorder arising from pathogenic variants in PRPS1, which encodes phosphoribosyl pyrophosphate synthetase 1-an enzyme essential for de novo nucleotide biosynthesis. Affected individuals typically exhibit sensorineural hearing loss, intellectual disability, cerebellar ataxia, and recurrent infections. However, despite the severity of these clinical manifestations, therapeutic interventions remain limited, largely due to an incomplete understanding of the cellular pathophysiology underlying the disorder. In this study, we generated patient-specific induced pluripotent stem cells harboring the PRPS1 p.V42L variant and differentiated them into neural stem cells (NSCs) and neurons to elucidate disease mechanisms and explore potential therapeutic strategies. Patient-derived NSCs demonstrated significantly reduced proliferative capacity, aberrant nuclear morphology, and increased neuronal senescence, while mitochondrial integrity and function were largely preserved. Neurons differentiated from these NSCs exhibited impaired neurite outgrowth and reduced branching complexity, indicative of disrupted neurodevelopmental processes. Notably, supplementation with nicotinamide mononucleotide, a precursor of nicotinamide adenine dinucleotide (NAD+), partially ameliorated defects in NSC proliferation, nuclear architecture, and neuronal morphology. Collectively, these findings delineate key cellular mechanisms underlying PRPS1-associated neurodevelopmental pathology and identify NAD+ metabolic augmentation as a promising therapeutic avenue for Arts syndrome and related PRPS1-mediated disorders.
Neurological Consequences of Infantile Vitamin B12 Deficiency - A Prospective Cohort Study.
The purpose of this research is to study the neurological consequences of infantile vitamin B12 deficiency on the developing brain. A prospective cohort study was done in consecutive children with Infantile B12 deficiency. Clinical evaluation, developmental assessment, blood investigations, and a magnetic resonance imaging (MRI) of the brain were performed at baseline and after therapy with injectable vitamin B12. Among 141 children (median age-13 months), developmental delay was observed in 131 (93%), and 79 (56%) had regression. Eighty (57%) babies had head circumference of < -2 Z score. At baseline, the MRI of the brain was abnormal in 137 (97.2%), showing thinning of corpus callosum (n = 133, 94.3%), cerebral cortical atrophy (n = 128, 90.8%), cerebellar atrophy (n = 126, 89.4%), atrophy of midbrain (n = 81, 57.4%) and pons (n = 78, 55.3%). A follow-up MRI done in 98 (69.5%) showed 66 (67%) had one or more residual abnormalities. The baseline full-scale developmental quotient was 22 (interquartile range: 13-30), while the follow-up full-scale developmental quotient score was 47.5 (interquartile range: 42.5-55). Seventy-nine (67.5%) had a follow-up developmental quotient of less than 50, implying moderate to severe developmental retardation. Despite therapy, children affected by the infantile B12 deficiency syndrome have significant lasting effects on the brain, evident as poor head growth, developmental deficits, and residual brain imaging changes.
Two siblings with CCDC32-related cardiofacioneurodevelopmental syndrome diagnosed by clinical RNA-sequencing and review of literature.
Cardiofacioneurodevelopmental syndrome (CFNDS, MIM:619123) is a rare genetic disorder caused by bi-allelic pathogenic variants in CCDC32. So far, CFNDS has only been described in four living individuals and one terminated fetus from four families, and the clinical phenotype can include microcephaly, facial malformations, developmental delay, cerebellar hypoplasia, and cardiac anomalies. We present a family with two affected individuals who were diagnosed through clinical RNA sequencing (RNA-seq) after conventional DNA diagnostics did not yield a molecular cause. Skipping of two exons in CCDC32 transcript was identified, consistent with a bi-allelic deletion including exons 3 and 4 of CCDC32. This deletion was not detected in previous SNP array analyses and trio exome sequencing focusing on genes related to intellectual disability and congenital malformations, highlighting the complementary value of RNA-seq. Furthermore, we review the clinical phenotype of this rare disorder and its potential disease mechanisms.
Publicações recentes
Tracheal cartilaginous sleeve prevalence in syndromic craniosynostosis: A single institution study.
Early posterior cranial vault distraction in syndromic craniosynostosis: orbital and ocular changes.
Generation of human induced pluripotent stem cell lines from patients with FGFR2-linked syndromic craniosynostosis.
Syndromic Craniosynostosis: The Hidden Burden of Comorbidities on Surgical Outcomes.
Determining the cause of optic nerve atrophy in syndromic craniosynostosis using logistic regression.
📚 EuropePMCmostrando 196
Znhit3 regulates p53/p21 signaling and governs cerebellar granule cell development.
Cell death and differentiationBiallelic Novel SKOR2 Variants in Individuals With Cerebellar Hypoplasia and Intellectual Disability, Expanding the Phenotypic Spectrum of Valence-Farazi Cerebellar Ataxia Syndrome.
American journal of medical genetics. Part AChild Neurology: Multiple Genetic Etiologies Causing Dandy-Walker Variant With Microcephaly, Epilepsy, and Global Developmental Delay.
NeurologyPRPS1 (p.V42L) Mutation in Arts Syndrome Induces Aberrant Neural Stem Cell Development and Neuronal Senescence-Like Phenotype: Rescue by Nicotinamide Mononucleotide Supplementation.
International journal of stem cellsErythropoietin alleviates syndrome-associated intellectual disability and autism-like behavior in Zbtb20-haploinsufficient Primrose syndrome mouse model.
JCI insightAltered Brain Structure in an ATRX-Deficient Mouse Model of Autism Spectrum Disorder.
Autism research : official journal of the International Society for Autism ResearchChromatin remodelling subunit SMARCB1 is implicated in dendrite development and complex brain functions.
Acta neuropathologica communicationsNeurological Consequences of Infantile Vitamin B12 Deficiency - A Prospective Cohort Study.
Pediatric neurologyFirst demyelinating attack in children: A twelve year single center cohort.
Multiple sclerosis and related disordersTwo siblings with CCDC32-related cardiofacioneurodevelopmental syndrome diagnosed by clinical RNA-sequencing and review of literature.
European journal of human genetics : EJHGClinical Phenotypes and Prognosis of Anti-mGluR1 Encephalitis: A Single-Center Case Series and Comprehensive Literature Review.
Diagnostics (Basel, Switzerland)Expanding the neurological spectrum of HTLV-1 beyond HAM/TSP: a contemporary perspective.
Lancet regional health. AmericasBiallelic TTBK1 variant causes a severe syndromic neurodevelopmental disorder: clinical and genetic insights from two siblings.
Journal of medical geneticsGenetic, Clinical and Neuroradiological Spectrum of MED-Related Disorders: An Updated Review.
GenesSpinocerebellar ataxia, autosomal recessive type 23 (SCAR23) with compound TDP2 variants: clinical, molecular, and quantitative follow-up.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology[Clinical and genetic analysis of a child with X-linked Hoyeraal-Hreidarsson syndrome due to variant of DKC1 gene and a literature review].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsCerebellar and cerebral contributions to dual physical-cognitive impairment in middle-aged and older adults: Potential biomarkers and therapeutic targets for healthy aging.
NeuroImageClinical Framework for Motor Rehabilitation in Parkinsonism: Integrating Individualized and Syndrome-Specific Approaches.
Brain & NeuroRehabilitationEffect of Aminopyridines on Oculomotor Dysfunction in Anti-GAD Ataxia: A Brief Report.
Cerebellum (London, England)Deciphering Spastic Ataxia: Clinical and Genetic Profiles.
Neurology. GeneticsVPS13D-Related Disorders: Description of New Variant and Phenotypic Spectrum Based on Age of Onset.
Cerebellum (London, England)Daflon attenuates cisplatin-induced cerebellar neurotoxicity, anxiety-like behavior, and motor dysfunction by downregulating TLR4/NF-kB signaling.
BMC pharmacology & toxicologyWDR81 Mutation in Two Siblings: A Case Report and Review of Literature.
Molecular syndromologyDiagnostic odyssey of opsoclonus-myoclonus syndrome and barriers to early detection.
Brain & developmentCase Report: A 1-year progression of mediolateral gait instability during tandem walking in FXTAS.
Frontiers in sports and active livingEPG5-Related Disorders in Seven New Patients: Refining the Phenotypic Spectrum and Insights on Phenotype-Genotype Correlations.
Journal of molecular neuroscience : MNADNP Exhibits Methyltransferase Activity in Overexpression Systems and Modulates DNA and Histone Methylation.
Autism research : official journal of the International Society for Autism ResearchA Novel Mouse Model for Developmental and Epileptic Encephalopathy by Purkinje Cell-Specific Deletion of Scn1b.
The Journal of neuroscience : the official journal of the Society for NeuroscienceA New Variant in the NALCN Channel Is Responsible for Cerebellar Ataxia and Cognitive Impairment.
GenesBridging Genotype to Phenotype in KMT5B-Related Syndrome: Evidence from RNA-Seq, 18FDG-PET, Clinical Deep Phenotyping in Two New Cases, and a Literature Review.
GenesMotor, cognitive, affective, and communication outcomes in patients with sustained remission of opsoclonus-myoclonus-ataxia syndrome.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyUnderstanding cognitive features of cervical dystonia: application of the cerebellar cognitive affective syndrome scale (CCAS-S).
Journal of neural transmission (Vienna, Austria : 1996)De Novo Heterozygous ZFX Frameshift Variant in a Female With an X-Linked Neurodevelopmental Disorder.
American journal of medical genetics. Part ACase 341: Infratentorial Posterior Reversible Encephalopathy Syndrome Associated with Interferon-β in Relapsing Multiple Sclerosis.
RadiologyLance-Adams Syndrome: An Updated Review of a Rare Post-Hypoxic Complication.
Tremor and other hyperkinetic movements (New York, N.Y.)Diagnostic Challenges in Fahr's Disease: A Rare Case of Extensive Basal Ganglia Calcifications.
CureusNeuropathological findings of very low-density lipoprotein receptor-related cerebellar hypoplasia in a full-term fetus.
Journal of neuropathology and experimental neurologyNovel neuropathological observations in an adult with Dravet syndrome.
EpilepsiaChronic Early-Life Obesity Linked to Childhood Impulsivity Predicts Long-Term Psychosis Trajectory Through Dose-Dependent Cerebellar Dysmaturation in 22q11.2 Deletion Syndrome.
Biological psychiatry. Cognitive neuroscience and neuroimagingDeletion of the SHORT Syndrome Gene Prkce Results in Brain Atrophy and Cognitive and Motor Behavior Deficits in Mice.
Neuroscience bulletinSpeech and Language Development of Two Brothers With Bainbridge-Ropers Syndrome: Phenotypic and Bioinformatic Support for a Cerebellar ASXL3 Hypothesis.
American journal of medical genetics. Part AIdentification of two novel pathogenic mutations in the SKOR2 gene linked to cerebellar hypoplasia and a broad spectrum of neurodevelopmental delay in two Iranian families.
Journal of human geneticsSpatial perspective taking is impaired in spinocerebellar ataxias and Friedreich ataxia.
Scientific reportsA comparative study on clinico-radiological profile, treatment responses and outcomes of double seronegative NMOSD compared to AQP4-IgG positive NMOSD, and MOGAD.
Multiple sclerosis and related disordersRAB23 loss-of-function mutation causes context-dependent ciliopathy in Carpenter syndrome.
PLoS geneticsNatural history of SPTBN4-related neurodevelopmental disorder with hypotonia, neuropathy, and deafness.
Orphanet journal of rare diseasesPoretti-Boltshauser Syndrome: A Potential Pathognomonic "Wolfjaw" Pattern of Retinal Perfusion.
Ophthalmic surgery, lasers & imaging retinaNeurological Complications in Inborn Errors of Immunity: A Scoping Review of Clinical Spectrum, Pathophysiological Mechanisms, and Therapeutic Strategies.
Clinical reviews in allergy & immunologyPrenatal and postnatal findings in cerebellofaciodental syndrome: a rare genetic disorder.
Clinical dysmorphologyExpanding the Clinical, Pathological, and Molecular Phenotypes of Tetratricopeptide 19 (TTC19) Gene Mutations: A Case Report from India.
Neurology IndiaFunctional Assessment of the Subjects with Unertan Syndrome: 10 Years Follow-Up Study.
Cerebellum (London, England)Developmental and Epileptic Encephalopathy as a Novel Clinical Hallmark of SCA21.
NeuropediatricsRitscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy.
Science translational medicineNeuropathy in GAA-FGF14 Late-Onset Cerebellar Ataxia (SCA27B): Prevalence and Characteristics.
European journal of neurologyExpansion of the Genotypic and Phenotypic Spectrum of TCTN3-Related Joubert Syndrome.
GenesCerebellum-Predominant Progressive Multifocal Leukoencephalopathy: An Under-recognized Cause of Cerebellar Syndrome among People with Human Immunodeficiency Virus Infection.
Neurology IndiaPOLR3A rare variants in a patient with intellectual disability, ataxic gait and cortical malformations: a case-report.
Italian journal of pediatricsCase Series of Prenatally Diagnosed Cri du Chat Syndrome With Associated Magnetic Resonance Imaging Findings.
Pediatric neurologyFrench guidelines for the diagnosis and management of pure hereditary spastic paraplegia.
Revue neurologiqueA case series of joubert syndrome evaluated with whole exome sequencing and the utility of optical genome mapping in the diagnosis.
NeurogeneticsAbnormal brain networks in Meiges syndrome based on centrality analysis and functional network connectivity: a cross-sectional analysis.
Brain imaging and behaviorPhenotypic Spectrum of KATNIP-Associated Joubert Syndrome: Possible Association with Esophageal Atresia and Review of the Literature.
GenesJun N-Terminal Kinase Inhibitor Suppresses CASK Deficiency-Induced Cerebellar Granular Cell Death in MICPCH Syndrome Model Mice.
CellsA 19q13 microdeletion syndrome presenting with punding, frangophilia, hypermetamorphosis, frontal lobe and vermal hypoplasia, with depression misdiagnosed as schizophrenia, treated with mirtazapine.
Psychiatric geneticsGlobally Reduced Brain Volume in Rett Syndrome.
Pediatric neurologyNeurological findings in a cohort of adults with down syndrome.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyNovel biallelic NUP107 variants affect the nuclear pore complex and expand the clinical spectrum to include brain malformations.
Journal of medical geneticsPediatric Opsoclonus-Myoclonus-Ataxia Syndrome can Lead to Long-Term Neurological, Neuropsychological, and Cognitive Sequelae Associated with Cerebellar Atrophy.
Cerebellum (London, England)Clinical Features and Outcomes of Glutamic Acid Decarboxylase-65 Antibody-Associated Pure Cerebellar Ataxia and Stiff Person Syndrome Spectrum Disorders: A Single-Center Cohort Study.
European journal of neurologyCore diagnostic features of stiff person syndrome: insights from a case-control study.
Journal of neurologyFurther Delineation of the AUTS2 HX Repeat Domain-Related Phenotype.
American journal of medical genetics. Part AExpanding the spectrum of ATP8A2 mutations: a new splicing variant and systematic review of CAMRQ4 syndrome.
Molecular biology reportsCerebellar Ataxia-deafness-narcolepsy (ADCA) syndrome. Description of a variable family phenotype.
Acta neurologica BelgicaDe Novo Splice-Site Variant in DKC1 in a Female With Clinical Features of Hoyeraal-Hreidarsson Syndrome.
American journal of medical genetics. Part ANeuroanatomical features of NAA10 and NAA15-related neurodevelopmental syndromes.
Journal of neuroradiology = Journal de neuroradiologieSensory Dysfunction in ALS and Other Motor Neuron Diseases: Clinical Relevance, Histopathology, Neurophysiology, and Insights from Neuroimaging.
BiomedicinesDiffuse but Non-homogeneous Brain Atrophy: Identification of Specific Brain Regions and Their Correlation with Clinical Severity in Rett Syndrome.
Brain & developmentMotor markers of congenital cerebellar hypoplasia.
NeuropsychologiaNatural history of patients with autosomal dominant WFS1 pathogenic variants associated with sensorineural hearing loss and optic atrophy.
medRxiv : the preprint server for health sciencesBiallelic variants in GTF3C3 encoding a subunit of the TFIIIC2 complex are associated with neurodevelopmental phenotypes in humans and zebrafish.
Brain communicationsAutistic behavior is a common outcome of biallelic disruption of PDZD8 in humans and mice.
Molecular autismCerebellar Ataxia, Impaired Intellectual Development, and Disequilibrium Syndrome-2: A Case Report.
CureusThe Clinical and Molecular Spectrum of Patients With X-Linked Intellectual Disability and Novel Variations in Different Genes.
Pediatric neurologyMovement Disorders in Hereditary Cerebellar Ataxia.
Movement disorders clinical practiceClinical, biological, and neuroimaging profiles for motoric cognitive risk syndrome in older adults: The MIND-China study.
Journal of internal medicineReevaluation of the Impact of the Novel Likely Pathogenic Variant c.1286_1288delAGA in the ATP8A2 Gene: A 7-Year Follow-Up With Clinical, Genetic, and ACMG Insights in an Iranian Family.
Molecular genetics & genomic medicinePoretti-Boltshauser Syndrome: A Report of Two Cases From Bahrain With a Novel Mutation and Literature Review.
CureusPhenotypic variability in two siblings with Poretti-Boltshauser syndrome.
Global medical geneticsSmartphone postural sway and pronator drift tests as measures of neurological disability.
BMC neurologyViral vector-mediated SLC9A6 gene replacement reduces cerebellar dysfunction in the shaker rat model of Christianson syndrome.
bioRxiv : the preprint server for biologyAzygos Vein Stenosis in Frontotemporal Dementia Sagging Brain Syndrome.
AJNR. American journal of neuroradiologyNeurodevelopmental Abnormalities in Down Syndrome: Assessing Structural and Functional Deficits.
CureusNovel Meningoencephalomyelitis Associated With Vimentin IgG Autoantibodies.
JAMA neurologyAn X-Linked Ataxia Syndrome in a Family with Hearing Loss Associated with a Novel Variant in the BCAP31 Gene.
Movement disorders : official journal of the Movement Disorder SocietyClinical Manifestations and Treatment Responses in Pediatric Neurofascin 155-IgG4 Autoimmune Nodopathy.
Neurology(R) neuroimmunology & neuroinflammationRORA-neurodevelopmental disorder: A unique triad of developmental disabilities, cerebellar anomalies, and myoclonic seizures.
Genetics in medicine : official journal of the American College of Medical GeneticsBrain volumes, cognitive, and adaptive skills in school-age children with Down syndrome.
Journal of neurodevelopmental disordersStructural and functional properties of the N- and C-terminal segments of the P4-ATPase phospholipid flippase ATP8A2.
The Journal of biological chemistryNon-malignant features of cancer predisposition syndromes manifesting in childhood and adolescence: a guide for the general pediatrician.
World journal of pediatrics : WJPTbx1 haploinsufficiency leads to local skull deformity, paraflocculus and flocculus dysplasia, and motor-learning deficit in 22q11.2 deletion syndrome.
Nature communicationsBiallelic variants in GTF3C3 result in an autosomal recessive disorder with intellectual disability.
Genetics in medicine : official journal of the American College of Medical GeneticsN-Acetylcysteine Counteracts Immune Dysfunction and Autism-Related Behaviors in the Shank3b Mouse Model of Autism Spectrum Disorder.
Antioxidants (Basel, Switzerland)Novel BRAT1 Deep Intronic Variant Affects Splicing Regulatory Elements Causing Cerebellar Hypoplasia Syndrome: Genotypic and Phenotypic Expansion.
Clinical geneticsExpanding the phenotyping spectrum of Witteveen Kolk syndrome: first report of generalized dystonia and cerebellar ataxia.
Acta neurologica BelgicaDe novo missense variants in the PP2A regulatory subunit PPP2R2B in a neurodevelopmental syndrome: potential links to mitochondrial dynamics and spinocerebellar ataxias.
Human molecular geneticsA Novel Compound Heterozygous Genotype of the WDR73 Gene Associated With a Psychomotor Retardation Syndrome Without Cerebellar Atrophy and Other CNS Structural Abnormalities.
Clinical geneticsPHARC syndrome: an overview.
Orphanet journal of rare diseasesRehabilitation and Physiotherapy Action Strategy for an Acute Case of Lateral Medullary Syndrome: A Case Report.
CureusPediatric Spinal Vascular Abnormalities: Overview, Diagnosis, and Management.
Neuroimaging clinics of North AmericaCACNA1A haploinsufficiency leads to reduced synaptic function and increased intrinsic excitability.
Brain : a journal of neurologyNew Insights Into TRMT10A Syndrome: Case Report and Literature Review.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsCase report: Neuroacanthocytosis associated with novel variants in the VPS13A gene with concomitant nucleotide expansion for CANVAS and assessment with osmotic gradient ektacytometry.
Frontiers in neuroscienceBiallelic missense CEP55 variants cause prenatal MARCH syndrome.
Journal of human geneticsCornelia de Lange Syndrome: Expanding the Neuropathological Spectrum and Clinical Correlations.
Fetal and pediatric pathologyConsolidating the Role of Mutated ATP2B2 in Neurodevelopmental and Cerebellar Pathologies.
Clinical geneticsAtlantoaxial Instability due to Os Odontoideum in a Child with Christianson Syndrome.
Molecular syndromology[Genetic analysis of a fetus with Coffin-Siris syndrome 2 due to a novel variant of ARID1A gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics[Cerebellar cognitive affective syndrome and vertebrobasilar ischemia. From cerebello-cerebral diaschisis to "dysmetria of thought"].
Psychiatrike = PsychiatrikiNeural precursor cells rescue symptoms of Rett syndrome by activation of the Interferon γ pathway.
EMBO molecular medicineClinical Course of Neurologic Adverse Events Associated With Immune Checkpoint Inhibitors: Focus on Chronic Toxicities.
Neurology(R) neuroimmunology & neuroinflammationThe overlapping of phenotypes in Wiedemann-Steiner, Kleefstra and Coffin-Siris syndromes: a study of eleven patients.
Italian journal of pediatricsCACNA1G Causes Dominantly Inherited Myoclonus-Ataxia with Intellectual Disability: A Case Report.
Cerebellum (London, England)Christianson syndrome across the lifespan: genetic mutations and longitudinal study in children, adolescents, and adults.
Journal of medical geneticsNerve ultrasound in CANVAS-spectrum disease: Reduced nerve size distinguishes genetically confirmed CANVAS from other axonal polyneuropathies.
Journal of the peripheral nervous system : JPNSDevelopmental, Cognitive, Ocular Motor, and Neuroimaging Findings Related to SUFU Haploinsufficiency: Unraveling Subtle and Highly Variable Phenotypes.
Pediatric neurologyPhenotypic Spectrum and Natural History of Gillespie Syndrome. An Updated Literature Review with 2 New Cases.
Cerebellum (London, England)Immune Checkpoint Inhibitor-Related Cerebellar Toxicity: Clinical Features and Comparison with Paraneoplastic Cerebellar Ataxia.
Cerebellum (London, England)Natural history of non-polyglutamine CACNA1A disease in Austria.
Journal of neurologyExome sequencing confirms the clinical diagnosis of both joubert syndrome and klinefelter syndrome with keratoconus in a han Chinese family.
Frontiers in geneticsSRPK3 Is Essential for Cognitive and Ocular Development in Humans and Zebrafish, Explaining X-Linked Intellectual Disability.
Annals of neurologyA novel missense variant in the ATPase domain of ATP8A2 and review of phenotypic variability of ATP8A2-related disorders caused by missense changes.
NeurogeneticsDiagnostic MRI Score to Differentiate Susac Syndrome from Primary Angiitis of the Central Nervous System and Multiple Sclerosis.
Annals of neurologyPrenatal diagnosis of a 15q24.1 microdeletion in a fetus with cerebral and urogenital abnormalities.
Clinical geneticsIdentification of novel variants in BRF1 gene from patient with developmental delay, hearing abnormality, and nervous system anomalies.
International journal of developmental neuroscience : the official journal of the International Society for Developmental NeuroscienceNeuroanatomical Features of NAA10- and NAA15-Related Neurodevelopmental Syndromes.
medRxiv : the preprint server for health sciencesDe novo and inherited monoallelic variants in TUBA4A cause ataxia and spasticity.
Brain : a journal of neurologyMicrocornea, cerebellar hypoplasia and hyperlax joints-unusual combo in rare Ehlers-Danlos syndrome-musculocontractural type 1.
BMJ case reportsCASK pathogenic variant which expands the clinical spectrum for MICPCH syndrome in an adult patient.
American journal of medical genetics. Part AExpanding the clinical phenotype and genetic spectrum of GEMIN5 disorders: Early-infantile developmental and epileptic encephalopathies.
Brain and behaviorRescue of myocytes and locomotion through AAV2/9-2YF intracisternal gene therapy in a rat model of creatine transporter deficiency.
Molecular therapy. Methods & clinical developmentLance-Adams Syndrome in the Intensive Care Unit: A Case Report.
CureusStructural deviations of the posterior fossa and the cerebellum and their cognitive links in a neurodevelopmental deletion syndrome.
Molecular psychiatryBedside clinical assessment of patients with common upper limb tremor and algorithmic approach.
Asian biomedicine : research, reviews and newsZmiz1 is a novel regulator of brain development associated with autism and intellectual disability.
Frontiers in psychiatryADNP dysregulates methylation and mitochondrial gene expression in the cerebellum of a Helsmoortel-Van der Aa syndrome autopsy case.
Acta neuropathologica communicationsClinical, Neuroimaging, and Metabolic Footprint of the Neurodevelopmental Disorder Caused by Monoallelic HK1 Variants.
Neurology. GeneticsClinical and Molecular Spectrum of Autosomal Recessive CA8-Related Cerebellar Ataxia.
Movement disorders : official journal of the Movement Disorder SocietyJoubert syndrome-derived induced pluripotent stem cells show altered neuronal differentiation in vitro.
Cell and tissue researchLoss-of-function variant in spermidine/spermine N1-acetyl transferase like 1 (SATL1) gene as an underlying cause of autism spectrum disorder.
Scientific reportsVestibular Testing and Impairments in Postoperative Pediatric Cerebellar Mutism Syndrome: A Case Series.
Pediatric neurologyFunctional and in silico analysis of ATP8A2 and other P4-ATPase variants associated with human genetic diseases.
Disease models & mechanismsCase Report: An Adult Case of Poretti-Boltshauser Syndrome Diagnosed by Medical Checkup.
Cerebellum (London, England)Measurement of synaptic density in Down syndrome using PET imaging: a pilot study.
Scientific reportsNovel genotype-phenotype correlations, differential cerebellar allele-specific methylation, and a common origin of the (ATTTC)n insertion in spinocerebellar ataxia type 37.
Human geneticsPGAP2-Related Hyperphosphatasia-Mental Retardation Syndrome: Report of a Novel Patient, Toward a Broadening of Phenotypic Spectrum and Therapeutic Perspectives.
NeuropediatricsCerebellar cognitive affective syndrome with long-term features of autism spectrum disorder: evidence in a 9-year-old girl after vermian medulloblastoma surgery.
Child neuropsychology : a journal on normal and abnormal development in childhood and adolescenceUnderstanding the role of AMPA receptors in autism: insights from circuit and synapse dysfunction.
Frontiers in psychiatryA novel ACTB variant in an atypical case of Baraitser-Winter syndrome with cerebellar hypoplasia and diaphragmatic hernia.
Clinical dysmorphologyCerebellar Heterotopia: Broadening the Neuroradiological Spectrum of KBG Syndrome.
Cerebellum (London, England)Investigating brain alterations in the Dp1Tyb mouse model of Down syndrome.
Neurobiology of diseaseA genetic variant in the MAST1 gene is associated with mega-corpus-callosum syndrome with hypoplastic cerebellar vermis, in a fetus.
Molecular genetics & genomic medicineThe effects of response disequilibrium on social media use: A laboratory analogue.
Behavioural processesIRF2BPL Causes Mild Intellectual Disability Followed by Late-Onset Ataxia.
Neurology. GeneticsRole of the repeat expansion size in predicting age of onset and severity in RFC1 disease.
Brain : a journal of neurologyMultiple sclerosis under the age of ten: the challenge of a rare diagnosis in a special population - a case series.
Frontiers in neuroscienceAbnormal Neurologic Findings in Patients With Sickle Cell Disease Without a History of Major Neurologic Events.
Neurology. Clinical practiceCraniocervical instability in patients with Ehlers-Danlos syndromes: outcomes analysis following occipito-cervical fusion.
Neurosurgical reviewLate-onset stiff-person syndrome: challenges in diagnosis and management.
Therapeutic advances in neurological disordersCoenzyme Q10: A Biomarker in the Differential Diagnosis of Parkinsonian Syndromes.
Antioxidants (Basel, Switzerland)Untangling adaptive functioning of PMM2-CDG across age and its impact on parental stress: a cross-sectional study.
Scientific reportsA novel NONO nonsense variant in a fetus with renal abnormalities.
Prenatal diagnosisCompound Heterozygosity in Cerebellar Ataxia, Mental Retardation, and Disequilibrium Syndrome Type 4.
Prilozi (Makedonska akademija na naukite i umetnostite. Oddelenie za medicinski nauki)Expanding clinical profiles and prognostic markers in stiff person syndrome spectrum disorders.
Journal of neurologyA case report of anti-GAD65 antibody-positive autoimmune encephalitis in children associated with autoimmune polyendocrine syndrome type-II and literature review.
Frontiers in immunologySplicing variant of WDR37 in a case of Neurooculocardiogenitourinary syndrome.
Brain & developmentNeurological disease in xeroderma pigmentosum: prospective cohort study of its features and progression.
Brain : a journal of neurologyAtaxia Syndrome With Hearing Loss and Nephronophthisis Associated With a Novel Homozygous Variant in XPNPEP3.
Neurology. GeneticsGenetic Insights into the causal relationship between cannabis use and diabetic phenotypes: A genetic correlation and Mendelian randomization study.
Drug and alcohol dependenceChristianson Syndrome across the Lifespan: An International Longitudinal Study in Children, Adolescents, and Adults.
medRxiv : the preprint server for health sciencesDiversity and Surgical Management of Intracranial Fungal Infections.
The Journal of craniofacial surgeryDetailed Analysis of ITPR1 Missense Variants Guides Diagnostics and Therapeutic Design.
Movement disorders : official journal of the Movement Disorder SocietyBi-allelic ACBD6 variants lead to a neurodevelopmental syndrome with progressive and complex movement disorders.
Brain : a journal of neurologyNanopore long-read sequencing analysis reveals ZIC1 dysregulation caused by a de novo 3q inversion with a breakpoint located 7 kb downstream of ZIC1.
Journal of human geneticsUndifferentiated psychosis or schizophrenia associated with vermis-predominant cerebellar hypoplasia.
American journal of medical genetics. Part APhenotypic variability in Joubert syndrome is partially explained by ciliary pathophysiology.
Annals of human geneticsMonocular Torsional Oscillopsia in Dentato-olivary Disconnection.
Cerebellum (London, England)Two novel cases of biallelic SMPD4 variants with brain structural abnormalities.
NeurogeneticsCompound heterozygous mutations in the helicase RTEL1 causing Hoyeraal-Hreidarsson syndrome with Blake`s pouch cyst: a case report.
The Turkish journal of pediatricsPanoramic variation analysis of a family with neurodevelopmental disorders caused by biallelic loss-of-function variants in TMEM141, DDHD2, and LHFPL5.
Frontiers of medicineAdvanced Optical Microscopy: Unveiling Functional Insights Regarding a Novel PPP2R1A Variant and Its Unreported Phenotype.
International journal of molecular sciencesGrey-matter correlates of empathy in 4-Repeat Tauopathies.
NPJ Parkinson's diseaseAssessment of Multiple Aspects of Upper Extremity Function Independent From Ambulation in Patients With Multiple Sclerosis.
International journal of MS careATP2B2 de novo variants as a cause of variable neurodevelopmental disorders that feature dystonia, ataxia, intellectual disability, behavioral symptoms, and seizures.
Genetics in medicine : official journal of the American College of Medical GeneticsAssessment and tailored physical rehabilitation approaches in persons with cerebellar impairments targeting mobility and walking according to the International Classification of Functioning: a systematic review of case-reports and case-series.
Disability and rehabilitationClinical and genetic characterization of a Chinese family with pontocerebellar hypoplasia type 7.
American journal of medical genetics. Part ANeuroimaging research in Williams syndrome: Beginning to bridge the gap with clinical care.
Neuroscience and biobehavioral reviewsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de craniossinostose-anomalias esqueléticas e cerebelares-dificuldades de aprendizagem.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de craniossinostose-anomalias esqueléticas e cerebelares-dificuldades de aprendizagem
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Znhit3 regulates p53/p21 signaling and governs cerebellar granule cell development.
- Child Neurology: Multiple Genetic Etiologies Causing Dandy-Walker Variant With Microcephaly, Epilepsy, and Global Developmental Delay.
- PRPS1 (p.V42L) Mutation in Arts Syndrome Induces Aberrant Neural Stem Cell Development and Neuronal Senescence-Like Phenotype: Rescue by Nicotinamide Mononucleotide Supplementation.
- Neurological Consequences of Infantile Vitamin B12 Deficiency - A Prospective Cohort Study.
- Two siblings with CCDC32-related cardiofacioneurodevelopmental syndrome diagnosed by clinical RNA-sequencing and review of literature.
- Tracheal cartilaginous sleeve prevalence in syndromic craniosynostosis: A single institution study.
- Early posterior cranial vault distraction in syndromic craniosynostosis: orbital and ocular changes.
- Generation of human induced pluripotent stem cell lines from patients with FGFR2-linked syndromic craniosynostosis.
- Syndromic Craniosynostosis: The Hidden Burden of Comorbidities on Surgical Outcomes.
- Determining the cause of optic nerve atrophy in syndromic craniosynostosis using logistic regression.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:672985(Orphanet)
- OMIM OMIM:616602(OMIM)
- MONDO:0014705(MONDO)
- GARD:18048(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
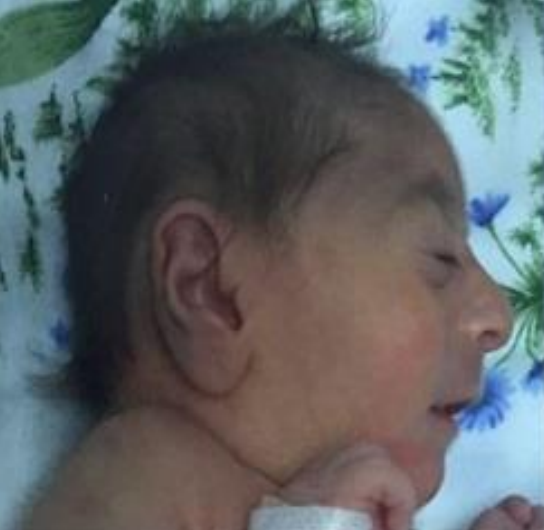
Síndrome de craniossinostose-anomalias esqueléticas e cerebelares-dificuldades de aprendizagem
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)