Síndrome epiléptica rara com início em recém-nascidos e lactentes, caracterizada por crises epilépticas severas e de difícil controle, frequentemente associadas a atraso no desenvolvimento neurológico e outras comorbidades. O diagnóstico precoce e o manejo multidisciplinar são cruciais para otimizar o prognóstico.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome epiléptico de início neonatal-infantil é uma condição rara caracterizada por crises epilépticas que começam nos primeiros meses de vida, durante o período neonatal ou na primeira infância. Esta página foi criada para oferecer informações acolhedoras e baseadas em evidências científicas para pacientes, familiares e cuidadores.[1][2]
Sinais e sintomas
Os sinais e sintomas associados a esta síndrome podem variar amplamente entre os indivíduos. Entre os achados mais frequentes estão: hipotonia (fraqueza muscular) no lactente, dificuldade de sucção, apneia (pausas na respiração), ataxia episódica (falta de coordenação motora que surge em crises), coreia (movimentos involuntários e rápidos), estrabismo (desalinhamento dos olhos) e comportamento autolesivo. Também podem ocorrer alterações estruturais como ponte nasal deprimida, defeito do septo ventricular (comunicação entre as câmaras do coração), hérnia umbilical, ureterocele (dilatação na porção final do ureter), falanges largas dos dedos dos pés, dedos curtos e largos, unha do polegar ausente, cifose (curvatura anormal da coluna), paquigiria (espessamento anormal do córtex cerebral) e atrofia cerebral difusa. Outros sintomas incluem déficit de crescimento, puberdade precoce, hiperatividade, anormalidades do sono, infecções respiratórias recorrentes (especialmente do trato respiratório inferior) e anormalidades difusas da substância branca do cérebro.[1][3]
Causas genéticas
A Síndrome epiléptico de início neonatal-infantil é uma condição genética, mas até o momento não há informações específicas sobre os genes envolvidos ou o padrão de herança disponíveis nas fontes oficiais consultadas. Pesquisas estão em andamento para identificar as bases genéticas exatas desta síndrome.[1][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica detalhada, incluindo a observação dos sintomas característicos e a exclusão de outras causas de epilepsia neonatal e infantil. Exames de imagem cerebral (como ressonância magnética) podem revelar achados como atrofia cerebral difusa ou paquigiria. Testes genéticos podem ser úteis para confirmar o diagnóstico, embora não haja informações específicas sobre variantes genéticas disponíveis nas fontes oficiais atuais.[1][4]
Tratamento e manejo
O tratamento é individualizado e deve ser conduzido por uma equipe multidisciplinar, incluindo neurologista, pediatra, geneticista e outros especialistas conforme necessário. O manejo das crises epilépticas pode incluir o uso de medicamentos anticonvulsivantes, mas é importante ressaltar que este texto não substitui a orientação médica. Cada paciente deve ter seu plano terapêutico definido pelo médico assistente. No Brasil, o acesso a alguns tratamentos pode ser parcialmente coberto pelo Sistema Único de Saúde (SUS), conforme a classificação de cobertura parcial.[1]
Tratamentos citados na literatura
A literatura científica menciona diversos fármacos que foram estudados ou associados a esta síndrome, mas é fundamental destacar que estas são associações mineradas automaticamente de bases de dados científicas (PubTator3) e NÃO representam recomendações de tratamento. Cada medicamento deve ser avaliado pelo médico responsável. Os fármacos citados incluem: aminohydroxybutyric acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic acid, valpromide e zonisamide. O número de publicações científicas associadas a cada um varia e não está disponível nesta base.[1]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e a resposta ao tratamento. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para melhorar a qualidade de vida. O diagnóstico precoce e o manejo adequado das crises epilépticas e dos sintomas associados podem contribuir para um melhor desenvolvimento e bem-estar.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome epiléptica rara com início em recém-nascidos e lactentes, caracterizada por crises epilépticas severas e de difícil controle, frequentemente associadas a atraso no desenvolvimento neurológico e outras comorbidades. O diagnóstico precoce e o manejo multidisciplinar são cruciais para otimizar o prognóstico.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome epiléptico de início neonatal-infantil é uma condição rara caracterizada por crises epilépticas que começam nos primeiros meses de vida, durante o período neonatal ou na primeira infância. Esta página foi criada para oferecer informações acolhedoras e baseadas em evidências científicas para pacientes, familiares e cuidadores.[1][2]
Sinais e sintomas
Os sinais e sintomas associados a esta síndrome podem variar amplamente entre os indivíduos. Entre os achados mais frequentes estão: hipotonia (fraqueza muscular) no lactente, dificuldade de sucção, apneia (pausas na respiração), ataxia episódica (falta de coordenação motora que surge em crises), coreia (movimentos involuntários e rápidos), estrabismo (desalinhamento dos olhos) e comportamento autolesivo. Também podem ocorrer alterações estruturais como ponte nasal deprimida, defeito do septo ventricular (comunicação entre as câmaras do coração), hérnia umbilical, ureterocele (dilatação na porção final do ureter), falanges largas dos dedos dos pés, dedos curtos e largos, unha do polegar ausente, cifose (curvatura anormal da coluna), paquigiria (espessamento anormal do córtex cerebral) e atrofia cerebral difusa. Outros sintomas incluem déficit de crescimento, puberdade precoce, hiperatividade, anormalidades do sono, infecções respiratórias recorrentes (especialmente do trato respiratório inferior) e anormalidades difusas da substância branca do cérebro.[1][3]
Causas genéticas
A Síndrome epiléptico de início neonatal-infantil é uma condição genética, mas até o momento não há informações específicas sobre os genes envolvidos ou o padrão de herança disponíveis nas fontes oficiais consultadas. Pesquisas estão em andamento para identificar as bases genéticas exatas desta síndrome.[1][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica detalhada, incluindo a observação dos sintomas característicos e a exclusão de outras causas de epilepsia neonatal e infantil. Exames de imagem cerebral (como ressonância magnética) podem revelar achados como atrofia cerebral difusa ou paquigiria. Testes genéticos podem ser úteis para confirmar o diagnóstico, embora não haja informações específicas sobre variantes genéticas disponíveis nas fontes oficiais atuais.[1][4]
Tratamento e manejo
O tratamento é individualizado e deve ser conduzido por uma equipe multidisciplinar, incluindo neurologista, pediatra, geneticista e outros especialistas conforme necessário. O manejo das crises epilépticas pode incluir o uso de medicamentos anticonvulsivantes, mas é importante ressaltar que este texto não substitui a orientação médica. Cada paciente deve ter seu plano terapêutico definido pelo médico assistente. No Brasil, o acesso a alguns tratamentos pode ser parcialmente coberto pelo Sistema Único de Saúde (SUS), conforme a classificação de cobertura parcial.[1]
Tratamentos citados na literatura
A literatura científica menciona diversos fármacos que foram estudados ou associados a esta síndrome, mas é fundamental destacar que estas são associações mineradas automaticamente de bases de dados científicas (PubTator3) e NÃO representam recomendações de tratamento. Cada medicamento deve ser avaliado pelo médico responsável. Os fármacos citados incluem: aminohydroxybutyric acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic acid, valpromide e zonisamide. O número de publicações científicas associadas a cada um varia e não está disponível nesta base.[1]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e a resposta ao tratamento. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para melhorar a qualidade de vida. O diagnóstico precoce e o manejo adequado das crises epilépticas e dos sintomas associados podem contribuir para um melhor desenvolvimento e bem-estar.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 60 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 174 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome epiléptico de início neonatal-infantil
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Epilepsy-Associated Variants of a Single SCN1A Codon Exhibit Divergent Functional Properties.
Pathogenic variants in SCN1A, which encodes the voltage-gated sodium channel NaV1.1, are associated with multiple epilepsy syndromes exhibiting a range of clinical severity. SCN1A variants are reported in different syndromes, including Dravet syndrome, which is associated with loss-of-function, whereas neonatal/infantile-onset developmental and epileptic encephalopathy (DEE) is associated with gain-of-function. Strategies to predict SCN1A variant pathogenicity and dysfunction have been proposed but are limited by available training data. We investigated the functional properties of four epilepsy-associated SCN1A variants affecting the same codon and sought to correlate channel dysfunction with phenotype. Whole-cell manual patch-clamp recording was performed on heterologously expressed NaV1.1 variants. Structural modeling of NaV1.1 variant proteins was conducted using AlphaFold 3. We describe an individual with early infantile-onset DEE associated with SCN1A-I1347T, and identified three additional cases from the literature or ClinVar with distinct variations of the same codon (I1347N, I1347V, I1347F). Functional studies demonstrated mixed function properties for I1347T, I1347V, and I1347F, but complete loss-of-function for I1347N. Structural models suggest important interactions between isoleucine-1347 and the sixth transmembrane helices of domains 3 and 4 that are disrupted most significantly with asparagine replacement at this position (I1347N). Pathogenic variants in SCN1A involving the same codon can produce divergent functional effects. Our findings suggest that predicting specific functional effects of SCN1A variants should not rely heavily on position in the protein.
Oxcarbazepine may be an effective option for Chinese pediatric patients with self-limited focal epilepsy of neonatal/infantile onset: a retrospective cohort study.
The aim of this study was to evaluate the long-term follow-up data of Chinese children with self-limited focal epilepsy with neonatal/infantile onset (SeLFE) and to investigate the clinical features, genetic background and treatment outcomes of this type of epileptic syndrome. We conducted a retrospective cohort study of twenty-six children with SeLFE admitted to or followed by the Department of Pediatrics, Second Affiliated Hospital of Xi'an Jiaotong University from October 2011 to October 2021. Treatment decisions were based on the children's seizure semiology, frequency, economy, medication accessibility, allergies and other factors, and initial medications including levetiracetam, phenobarbital and oxcarbazepine. All children were followed up regularly in the outpatient clinic. The 26 children, 13 male and 13 female, were followed for a mean of 54.0 (49.0, 58.5) months. Trio whole-exome sequencing (WES) revealed no pathogenic genetic abnormalities in 16 children, and known pathological genes including PRRT2, SCN2A and KCNQ2 were detected in 10 children. Thirteen children (50.0%) achieved complete seizure control after first-line monotherapy. Among the 12 patients who failed to respond to the first monotherapy, 9 patients achieved a seizure free status with oxcarbazepine, which was used as the second-line monotherapy or as add-on therapy. One patient recovered spontaneously without treatment. Although SeLFE is often self-limited, this study showed that complete seizure control is not always achieved with initial medication therapy. Oxcarbazepine may be an effective option for the treatment of SeLFE.
Biallelic PTPMT1 variants disrupt cardiolipin metabolism and lead to a neurodevelopmental syndrome.
Primary mitochondrial diseases (PMDs) are among the most common inherited neurological disorders. They are caused by pathogenic variants in mitochondrial or nuclear DNA that disrupt mitochondrial structure and/or function, leading to impaired oxidative phosphorylation (OXPHOS). One emerging subcategory of PMDs involves defective phospholipid metabolism. Cardiolipin, the signature phospholipid of mitochondria, resides primarily in the inner mitochondrial membrane, where it is biosynthesized and remodelled via multiple enzymes and is fundamental to several aspects of mitochondrial biology. Genes that contribute to cardiolipin biosynthesis have recently been linked with PMD. However, the pathophysiological mechanisms that underpin human cardiolipin-related PMDs are not fully characterized. Here, we report six individuals, from three independent families, harbouring biallelic variants in PTPMT1, a mitochondrial tyrosine phosphatase required for de novo cardiolipin biosynthesis. All patients presented with a complex, neonatal/infantile onset neurological and neurodevelopmental syndrome comprising developmental delay, microcephaly, facial dysmorphism, epilepsy, spasticity, cerebellar ataxia and nystagmus, sensorineural hearing loss, optic atrophy and bulbar dysfunction. Brain MRI revealed a variable combination of corpus callosum thinning, cerebellar atrophy and white matter changes. Using patient-derived fibroblasts and skeletal muscle tissue, combined with cellular rescue experiments, we characterized the molecular defects associated with mutant PTPMT1 and confirmed the downstream pathogenic effects that loss of PTPMT1 has on mitochondrial structure and function. To further characterize the functional role of PTPMT1 in cardiolipin homeostasis, we created a ptpmt1 knockout zebrafish. This model had abnormalities in body size, developmental alterations, decreased total cardiolipin levels and OXPHOS deficiency. Together, these data indicate that loss of PTPMT1 function is associated with a new autosomal recessive PMD caused by impaired cardiolipin metabolism, highlighting the contribution of aberrant cardiolipin metabolism towards human disease and emphasizing the importance of normal cardiolipin homeostasis during neurodevelopment.
Epilepsy-associated Variants of a Single SCN1A Codon exhibit Divergent Functional Properties.
Pathogenic variants in SCN1A, which encodes the voltage gated sodium channel Na V 1.1, are associated with multiple epilepsy syndromes exhibiting a range of clinical severity. Loss or gain of function SCN1A variants are reported in different syndromes including Dravet syndrome, which is associated with loss-of-function whereas neonatal/infantile-onset developmental and epileptic encephalopathy (DEE) is associated with gain-of-function. Strategies to predict SCN1A variant pathogenicity and dysfunction have been proposed but are limited by available training data. We investigated the functional properties of four epilepsy-associated SCN1A variants affecting the same codon and sought to correlate channel dysfunction with phenotype. Whole-cell manual patch-clamp recording was performed on heterologously-expressed Na V 1.1 variants. Structural modeling of Na V 1.1 variant proteins was conducted using AlphaFold 3. We describe an individual with early infantile onset DEE associated with SCN1A-I1347T, and identified three additional cases from the literature or ClinVar with distinct variation of the same codon (I1347N, I1347V, I1347F). Functional studies demonstrated mixed gain and loss of function properties for I1347T, I1347V, and I1347F, but complete loss-of-function for I1347N. Structural models suggest important interactions between isoleucine-1347 and the sixth transmembrane helices of domains 3 and 4 that are disrupted most significantly with asparagine replacement at this position (I1347N). Pathogenic variants in SCN1A involving the same codon can produce divergent functional effects. Our findings suggest that predicting specific functional effects of SCN1A variants should not rely heavily on position in the protein.
Genetic etiologies with a large NGS panel in a monocentric cohort of 1000 patients with pediatric onset epilepsies.
Genetic testing is now included in the diagnostic assessment of childhood onset epilepsies. We evaluated the yield of a targeted next generation sequencing (TNGS) panel dedicated to pediatric epilepsies. We tested by TNGS panel 1000 consecutive patients presenting with childhood onset epilepsies and including mainly patients with early onset epilepsies (under 2 years, 61%). Causal variants were identified in 31% of patients, spanning 78 different genes. Patients with benign familial neonatal/infantile epilepsy (BFN/IS) exhibited the highest rate of positive findings (82%). Developmental and epileptic encephalopathies (DEEs) had a global diagnostic yield of 37%, with epilepsy of infancy with migrating focal seizures (EIMFSI) and Dravet syndrome (DS) presenting the highest yield in this group (78%) and early infantile DEE (EIDEE) laying next with a yield of 43%. The lowest rates of genetic diagnosis were observed in infantile epileptic spasms syndrome (IESS, 17%), epilepsy with myoclonic-atonic seizures (EMAtS, 19%), and DEE-SWAS (14%). Patients with GEFS+ had a yield of 16%. Among patients with developmental encephalopathies and refractory seizures with onset after 2 years, TNGS yielded a 33% diagnostic rate. Atypical absences yielded 16%, focal epilepsy yielded 18%, and generalized epilepsies with refractory seizures yielded 13%. These groups exhibited a high genetic heterogeneity. TNGS is an effective first-step genetic screening in patients with high diagnostic yields (BFN/IS, EIMFS, DS, EIDEE) and for epilepsy syndromes associated with one or a few major genes (BFN/IS, EIMFS, DS, GEFS+, DEE-SWAS). Whole exome or genome sequencing (WES/WGS) should be considered as a second step in these groups with a probably relevant Mendelian inheritance. WES/WGS could be proposed as first-tier analysis in patients with IESS, EMAtS, generalized or focal epilepsies refractory to ASMs, and developmental encephalopathies with seizure onset after 2 years. However, the lower diagnostic yield obtained in these groups may suggest a complex inheritance. This study emphasizes the importance of accurately identifying different types of epilepsy and epilepsy syndromes to improve genetic testing strategies. We suggest that a targeted gene panel can be a good first step for some genetic conditions, such as benign familial neonatal/infantile epilepsy, Dravet syndrome, and epilepsy of infancy with migrating focal seizures.
Publicações recentes
Biallelic LGI1 and ADAM23 variants cause hippocampal epileptic encephalopathy via the LGI1-ADAM22/23 pathway.
Genetic etiologies with a large NGS panel in a monocentric cohort of 1000 patients with pediatric onset epilepsies.
Neonatal/infantile-onset genetic epilepsies: The utility of genetic testing for molecular etiology-specific diagnosis concerning therapeutic implications.
Are SCN2A-related BFNISs also responsible for seizures in adulthood? A case report opens new scenarios.
Voltage-gated sodium channel epilepsies in a tertiary care center: Phenotypic spectrum with correlation to predicted functional effects.
📚 EuropePMCmostrando 22
Epilepsy-Associated Variants of a Single SCN1A Codon Exhibit Divergent Functional Properties.
Annals of clinical and translational neurologyGenetic etiologies with a large NGS panel in a monocentric cohort of 1000 patients with pediatric onset epilepsies.
Epilepsia openOxcarbazepine may be an effective option for Chinese pediatric patients with self-limited focal epilepsy of neonatal/infantile onset: a retrospective cohort study.
Frontiers in pediatricsBiallelic PTPMT1 variants disrupt cardiolipin metabolism and lead to a neurodevelopmental syndrome.
Brain : a journal of neurologyVoltage-gated sodium channel epilepsies in a tertiary care center: Phenotypic spectrum with correlation to predicted functional effects.
Epilepsy & behavior : E&BNeurodevelopmental outcomes in a cohort of Australian families with self-limited familial epilepsy of neonatal/infantile onset.
SeizureEfficacy and tolerability of oxcarbazepine in the treatment of focal epilepsy in neonates and infants under 3 months of age: A single-center retrospective analysis.
Epilepsy researchSCN2A-Related Epilepsy: The Phenotypic Spectrum, Treatment and Prognosis.
Frontiers in molecular neuroscienceSCN2A Pathogenic Variants and Epilepsy: Heterogeneous Clinical, Genetic and Diagnostic Features.
Brain sciences[Clinical and genetic spectrum of SCN2A gene associated epilepsy and episodic ataxia].
Zhonghua er ke za zhi = Chinese journal of pediatrics[Genotypes and clinical features of neonatal-onset genetic epilepsy in 141 patients].
Zhonghua er ke za zhi = Chinese journal of pediatricsEpilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy.
GenesFurther corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes.
Molecular medicine (Cambridge, Mass.)Novel SCN2A mutation in a family associated with juvenile-onset myoclonus: Case report.
MedicineHCN1 mutation spectrum: from neonatal epileptic encephalopathy to benign generalized epilepsy and beyond.
Brain : a journal of neurology[Phenotype study of SCN2A gene related epilepsy].
Zhonghua er ke za zhi = Chinese journal of pediatricsDynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy.
Proceedings of the National Academy of Sciences of the United States of AmericaEpilepsy Syndromes in Childhood.
Continuum (Minneapolis, Minn.)Infantile Epileptic Encephalopathy Associated With SCN2A Mutation Responsive to Oral Mexiletine.
Pediatric neurologyPhenotypic Variability from Benign Infantile Epilepsy to Ohtahara Syndrome Associated with a Novel Mutation in SCN2A.
Molecular syndromologyEpileptic patients with de novo STXBP1 mutations: Key clinical features based on 24 cases.
EpilepsiaSCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures.
NeurologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome epiléptico de início neonatal-infantil.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome epiléptico de início neonatal-infantil
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Epilepsy-Associated Variants of a Single SCN1A Codon Exhibit Divergent Functional Properties.
- Oxcarbazepine may be an effective option for Chinese pediatric patients with self-limited focal epilepsy of neonatal/infantile onset: a retrospective cohort study.
- Biallelic PTPMT1 variants disrupt cardiolipin metabolism and lead to a neurodevelopmental syndrome.
- Epilepsy-associated Variants of a Single SCN1A Codon exhibit Divergent Functional Properties.
- Genetic etiologies with a large NGS panel in a monocentric cohort of 1000 patients with pediatric onset epilepsies.
- Biallelic LGI1 and ADAM23 variants cause hippocampal epileptic encephalopathy via the LGI1-ADAM22/23 pathway.
- Neonatal/infantile-onset genetic epilepsies: The utility of genetic testing for molecular etiology-specific diagnosis concerning therapeutic implications.
- Are SCN2A-related BFNISs also responsible for seizures in adulthood? A case report opens new scenarios.
- Voltage-gated sodium channel epilepsies in a tertiary care center: Phenotypic spectrum with correlation to predicted functional effects.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:693802(Orphanet)
- MONDO:0100022(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
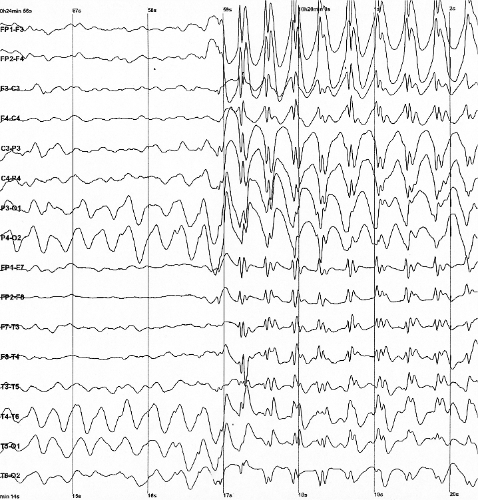
Síndrome epiléptico de início neonatal-infantil
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Reposicionamento
- fonte: Drug Repurposing Hub