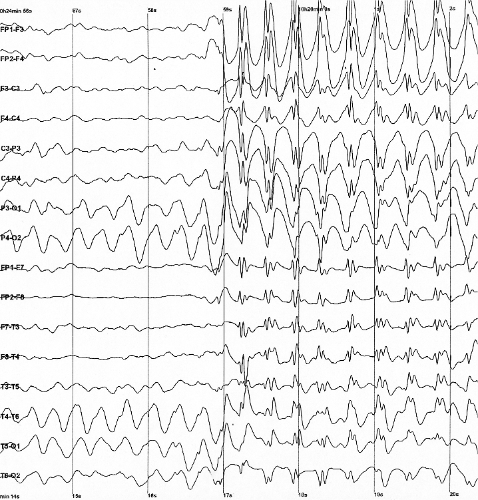
A epilepsia mioclônica familiar benigna do adulto (BAFME) é uma síndrome epiléptica hereditária caracterizada por tremores corticais nas mãos, espasmos mioclônicos e convulsões generalizadas ou focais ocasionais com um curso de doença não progressivo ou muito lentamente progressivo e sem sinais de demência precoce ou ataxia cerebelar.
Introdução
O que você precisa saber de cara
A epilepsia mioclônica familiar benigna do adulto (BAFME) é uma síndrome epiléptica hereditária caracterizada por tremores corticais nas mãos, espasmos mioclônicos e convulsões generalizadas ou focais ocasionais com um curso de doença não progressivo ou muito lentamente progressivo e sem sinais de demência precoce ou ataxia cerebelar.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 3 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 8 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
8 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Plays a role in RNA-mediated gene silencing by both micro-RNAs (miRNAs) and short interfering RNAs (siRNAs). Required for miRNA-dependent repression of translation and for siRNA-dependent endonucleolytic cleavage of complementary mRNAs by argonaute family proteins. As a scaffolding protein, associates with argonaute proteins bound to partially complementary mRNAs, and can simultaneously recruit CCR4-NOT and PAN deadenylase complexes
Cytoplasm, P-body
Epilepsy, familial adult myoclonic, 6
A form of familial myoclonic epilepsy, a neurologic disorder characterized by cortical hand tremors, myoclonic jerks and occasional generalized or focal seizures with a non-progressive or very slowly progressive disease course. Usually, myoclonic tremor is the presenting symptom, characterized by tremulous finger movements and myoclonic jerks of the limbs increased by action and posture. In a minority of patients, seizures are the presenting symptom. Some patients exhibit mild cognitive impairment. FAME6 inheritance is autosomal dominant.
In conjunction with another transmembrane protein, CNTNAP2, contributes to the organization of axonal domains at nodes of Ranvier by maintaining voltage-gated potassium channels at the juxtaparanodal region. May be involved in cell adhesion
Cell membrane
Epilepsy, early-onset, 5, with or without developmental delay
An autosomal recessive neurologic disorder characterized by a combination of various seizure types with onset in the first decade of life or during adolescence. Most patients have developmental delay, impaired intellectual development, and behavioral abnormalities.
Endoplasmic reticulum membrane-associated E3 ubiquitin ligase that plays a critical role in mitigating endoplasmic reticulum stress, the regulation of cholesterol and lipid homeostasis, and ferroptosis (PubMed:25088257, PubMed:35941365, PubMed:39216628). Acts as a pivotal component of both the Ac/N-degron pathway (targeting the N-terminal acetyl group of substrates) and the ER-associated protein degradation-cytosol (ERAD-C) pathway (targeting misfolded substrates) (PubMed:30425097, PubMed:359413
Endoplasmic reticulum membrane
Epilepsy, familial adult myoclonic, 3
A form of familial myoclonic epilepsy, a neurologic disorder characterized by cortical hand tremors, myoclonic jerks and occasional generalized or focal seizures with a non-progressive or very slowly progressive disease course. Usually, myoclonic tremor is the presenting symptom, characterized by tremulous finger movements and myoclonic jerks of the limbs increased by action and posture. In a minority of patients, seizures are the presenting symptom. Some patients exhibit mild cognitive impairment. FAME3 inheritance is autosomal dominant.
Epilepsy, familial adult myoclonic, 1
A form of familial myoclonic epilepsy, a neurologic disorder characterized by cortical hand tremors, myoclonic jerks and occasional generalized or focal seizures with a non-progressive or very slowly progressive disease course. Usually, myoclonic tremor is the presenting symptom, characterized by tremulous finger movements and myoclonic jerks of the limbs increased by action and posture. In a minority of patients, seizures are the presenting symptom. Some patients exhibit mild cognitive impairment. FAME1 inheritance is autosomal dominant.
Functions as a guanine nucleotide exchange factor (GEF), which activates Rap and Ras family of small GTPases by exchanging bound GDP for free GTP in a cAMP-dependent manner. Serves as a link between cell surface receptors and Rap/Ras GTPases in intracellular signaling cascades. Also acts as an effector for Rap1 by direct association with Rap1-GTP thereby leading to the amplification of Rap1-mediated signaling. Shows weak activity on HRAS. It is controversial whether RAPGEF2 binds cAMP and cGMP (
CytoplasmCytoplasm, perinuclear regionCell membraneLate endosomeCell junction
Epilepsy, familial adult myoclonic, 7
A form of familial myoclonic epilepsy, a neurologic disorder characterized by cortical hand tremors, myoclonic jerks and occasional generalized or focal seizures with a non-progressive or very slowly progressive disease course. Usually, myoclonic tremor is the presenting symptom, characterized by tremulous finger movements and myoclonic jerks of the limbs increased by action and posture. In a minority of patients, seizures are the presenting symptom. Some patients exhibit mild cognitive impairment. FAME7 inheritance is autosomal dominant.
Chromatin reader component of the ATAC complex, a complex with histone acetyltransferase activity on histones H3 and H4 (PubMed:18838386, PubMed:19103755, PubMed:27103431). YEATS2 specifically recognizes and binds histone H3 crotonylated at 'Lys-27' (H3K27cr) (PubMed:27103431). Crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors (PubMed:27103431)
Nucleus
Epilepsy, familial adult myoclonic, 4
A form of familial myoclonic epilepsy, a neurologic disorder characterized by cortical hand tremors, myoclonic jerks and occasional generalized or focal seizures with a non-progressive or very slowly progressive disease course. Usually, myoclonic tremor is the presenting symptom, characterized by tremulous finger movements and myoclonic jerks of the limbs increased by action and posture. In a minority of patients, seizures are the presenting symptom. Some patients exhibit mild cognitive impairment. FAME4 inheritance is autosomal dominant.
Has a critical role in neuronal development, particularly in the formation and/or maintenance of dendritic spines and synapses (PubMed:25807484). Involved in the regulation of Wnt signaling (PubMed:25807484). It probably acts on beta-catenin turnover, facilitating beta-catenin interaction with GSK3B, phosphorylation, ubiquitination and degradation (By similarity). Functions as a transcriptional activator when bound to ZBTB33 (By similarity). May be involved in neuronal cell adhesion and tissue m
NucleusCell junction, adherens junctionCell projection, dendritePerikaryon
Alpha-2 adrenergic receptors are G protein-coupled receptors for catecholamines that activate G(i/o) protein pathway, thereby promoting adenylyl cyclase inhibition, ERK1/2 stimulation, and voltage-gated calcium channels suppression (PubMed:11056163, PubMed:2164221, PubMed:2172775, PubMed:23105096, PubMed:26811329). Control a variety of physiological processes, such as regulation of blood pressure, lipolysis and insulin release (PubMed:2164221). The rank order of potency for agonists of ADRA2B is
Cell membrane
Epilepsy, familial adult myoclonic, 2
A form of familial myoclonic epilepsy, a neurologic disorder characterized by cortical hand tremors, myoclonic jerks and occasional generalized or focal seizures with a non-progressive or very slowly progressive disease course. Usually, myoclonic tremor is the presenting symptom, characterized by tremulous finger movements and myoclonic jerks of the limbs increased by action and posture. In a minority of patients, seizures are the presenting symptom. Some patients exhibit mild cognitive impairment. FAME2 inheritance is autosomal dominant.
Variantes genéticas (ClinVar)
201 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
34 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Epilepsia mioclônica do adulto familiar
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Embryonic spinocerebellar ataxia type 37 AUUUC repeat RNA causes neurodevelopmental defects in zebrafish.
Onset of many neurodegenerative and neuromuscular diseases usually starts in adulthood; however, recent advances point toward neurodevelopmental changes as drivers of late neurodegeneration. How early neuropathological features occur in these conditions remains unclear, which is critical for timely therapeutic intervention. Here, we provide evidence that neurodevelopmental axonal defects initiate a motor phenotype in a zebrafish model of spinocerebellar ataxia type 37 (SCA37), a degenerative hereditary condition caused by an ATTTC repeat in the DAB1 gene. We investigated neuronal defects triggered by the embryonic AUUUC repeat RNA and their effects later in life by transiently expressing this RNA in embryos and analyzing innervation and motor function. We found abnormalities in motor neuron axonal outgrowth and muscle innervation. We also discovered disrupted embryonic motor activity and reduced locomotor distance and velocity in late adult zebrafish, demonstrating motor impairment. Moreover, we showed that NOVA2 expression rescues axonal defects, indicating dysfunction of NOVA2-regulated neurodevelopmental processes. Overall, our results establish embryonic expression of the AUUUC repeat RNA as a driver of axonal and synaptic abnormalities, interfering with neuronal circuits and culminating in adult motor dysfunction.
Novel, complex configurations of the MARCHF6 repeat expansion associated with progressive myoclonic epilepsy and familial adult myoclonic epilepsy.
Repeat expansions are a known cause of progressive myoclonic epilepsy (PME) and familial adult myoclonic epilepsy (FAME). We hypothesized that PME and FAME may have an overlapping phenotypic spectrum and searched for pathogenic repeat expansions in 18 individuals from 15 families with later-onset PME or FAME. We generated whole genome sequencing data by short-read sequencing and searched for known and novel repeat expansions. No known, pathogenic repeat expansions were identified. Instead, we discovered a novel TTGTA expansion in the gene MARCHF6 at the same location as the known, pathogenic FAME3 expansion in a PME family. Targeted long-read sequencing of this locus revealed a large, complex repeat structure harbouring an expansion of the pathogenic TTTCA repeat that causes FAME, surrounded by TTTTA and TTGTA expansions. Motivated by this discovery, we developed a new bioinformatic approach, mixSTR, to search for evidence of such complex expansions and discovered an additional novel configuration of the FAME3 expansion containing hidden pathogenic TTTCA expansions embedded within a TTTTA expansion in a second family clinically diagnosed with FAME. Both families had initially tested negative for the FAME3 expansion with standard RP-PCR and short-read genome sequencing analysis. We searched large epilepsy and population cohorts but did not identify any additional new individuals with complex FAME3 expansions. These findings have two important implications. Firstly, known repeat expansion loci with unusual repeat expansions, even if not known to be pathogenic, warrant further investigation as they may contain hidden pathogenic repeat expansions. Secondly, they provide molecular support for the clinical idea that PME and FAME have an overlapping phenotypic spectrum, and that the known FAME repeat expansions should be part of the diagnostic assessment of unsolved PMEs.
[Molecular genetics of benign adult familial myoclonus epilepsy].
Benign adult familial myoclonus epilepsy (BAFME) is an autosomal dominantly inherited disease characterized by infrequent seizures and tremorous myoclonus. The disease is also called familial adult myoclonic epilepsy (FAME) or familial cortical myoclonic tremor with epilepsy (FCMTE). Although the causes of BAFME had been unknown for a long, we identified TTTCA and TTTTA repeat expansions in intron 4 of SAMD12 as a cause of BAFME type 1. We also found TTTCA and TTTTA repeat expansions in TNRC6A and RAPGEF2 also cause the disease (BAFME types 6 and 7, respectively), thus proposing a concept of repeat motif-phenotype correlation. After that, TTTCA and TTTTA repeat expansions in STARD7, MARCHF6, YEATS2, and RAI1 have been identified as causes of BAFME types 2, 3, 4, and 8. The findings further supported the concept. The involvement of RNA-mediated toxicity, particularly of UUUCA repeats, is assumed to be the pathomechanism of this disease. The next step will be understanding the molecular pathomechanism of BAFME and identifying molecular targets of more efficient therapeutic approaches.
Targeted nanopore long-read sequencing panel for the molecular diagnosis of intronic expansion in familial adult myoclonic epilepsy.
Familial adult myoclonic epilepsy (FAME), an autosomal dominant disorder, is characterized by cortical myoclonus and occasional generalized tonic-clonic seizures. To date, intronic pentanucleotide repeat expansions in at least seven genes, including SAMD12, TNRC6A, YEATS2, MARCHF6, STARD7, RAPGEF2, and RAI1, have been reported as causative. Detecting these repeat expansions using conventional sequencing techniques (Sanger or short-read next-generation sequencing) is not feasible as they cannot reliably span or characterize long repetitive elements. Although genetic testing has been performed in some research laboratories, comprehensive long read-based panel is unavailable for clinical application. To address this gap, we developed a targeted long-read sequencing panel and applied it in a clinical diagnostic context for the first time. We designed a custom long-read sequencing panel targeting all seven known FAME-associated repeat loci using Oxford Nanopore Cas9-enrichment technology and applied it to a 47-year-old woman with familial cortical myoclonic tremor, clinically suspected to have FAME. The panel functioned as intended, providing robust on-target coverage across all loci, facilitating confident interrogation of each repeat region. At the SAMD12 locus, strand-aware histograms and read-level inspection demonstrated a clear pathogenic expansion, encompassing mixed TTTTA/TTTCA motifs with detectable TTTGA interruptions, consistent with FAME1. Using the crude allele prediction option of tandem-genotypes, the expanded allele contained approximately 689 additional repeats relative to the reference genome. The other six loci showed no pathogenic expansions. This targeted long-read panel enabled the first clinical molecular diagnosis of FAME using a comprehensive assay, yielding allele-resolved characterization of the pathogenic repeat and its motif composition. With further validation, this approach may serve as a clinically practical tool for reliable detection of FAME1 and for broader screening of other FAME subtypes, potentially reducing reliance on prolonged clinical observation or specialized electrophysiological testing.
First clinical diagnosis of FAME3 via commercial Long-Read sequencing reveals mosaic repeat expansion in MARCHF6 gene.
Familial Adult Myoclonic Epilepsy type 3 (FAME3) is a rare autosomal dominant disorder characterized by cortical tremor and epilepsy, caused by a noncoding pentanucleotide repeat expansion (TTTTA/TTTCA)n in the MARCHF6 gene. Conventional genetic testing often fails to detect this expansion due to its repetitive structure and intronic location. We evaluated a 61-year-old woman with refractory myoclonic and generalized tonic-clonic seizures, whose prior genetic testing-including exome and genome sequencing-was non-diagnostic. Using PacBio HiFi long-read whole-genome sequencing and the tandem repeat genotyping tool TRGT, we identified a pathogenic MARCHF6 intronic expansion. The proband harbored one allele with 15 TTTTA repeats and a second allele with a compound expansion of 661 TTTTA and 12 TTTCA repeats. Three affected relatives shared similarly expanded alleles, but with increasing repeat size in the latter generations. Importantly, analysis using TRGT-instability revealed repeat mosaicism in all affected individuals, reflected by variability in motif counts across individual sequencing reads. This somatic heterogeneity may contribute to the phenotypic penetrance, variable expressivity and pleiotropism seen in FAME3 disease expression. To our knowledge, this is the first clinical diagnosis of FAME3 using a commercially available long-read sequencing platform, underscoring its diagnostic utility in resolving complex repeat expansion disorders and uncovering biologically relevant mosaicism.
Publicações recentes
Embryonic spinocerebellar ataxia type 37 AUUUC repeat RNA causes neurodevelopmental defects in zebrafish.
Novel, complex configurations of the MARCHF6 repeat expansion associated with progressive myoclonic epilepsy and familial adult myoclonic epilepsy.
Targeted nanopore long-read sequencing panel for the molecular diagnosis of intronic expansion in familial adult myoclonic epilepsy.
First clinical diagnosis of FAME3 via commercial Long-Read sequencing reveals mosaic repeat expansion in MARCHF6 gene.
[Molecular genetics of benign adult familial myoclonus epilepsy].
📚 EuropePMC17 artigos no totalmostrando 24
Embryonic spinocerebellar ataxia type 37 AUUUC repeat RNA causes neurodevelopmental defects in zebrafish.
Disease models & mechanismsNovel, complex configurations of the MARCHF6 repeat expansion associated with progressive myoclonic epilepsy and familial adult myoclonic epilepsy.
Brain communicationsTargeted nanopore long-read sequencing panel for the molecular diagnosis of intronic expansion in familial adult myoclonic epilepsy.
BMC medical genomicsFirst clinical diagnosis of FAME3 via commercial Long-Read sequencing reveals mosaic repeat expansion in MARCHF6 gene.
Neurogenetics[Molecular genetics of benign adult familial myoclonus epilepsy].
Rinsho shinkeigaku = Clinical neurologySAMD12 as a Master Regulator of MAP4Ks by Decoupling Kinases From the CNKSR2 Scaffold.
Journal of molecular biologyGene-gene interaction network analysis indicates CNTN2 is a candidate gene for idiopathic generalized epilepsy.
NeurogeneticsDimeric structures of DNA ATTTC repeats promoted by divalent cations.
Nucleic acids researchFAME4-associating YEATS2 knockdown impairs dopaminergic synaptic integrity and leads to seizure-like behaviours in Drosophila melanogaster.
Progress in neurobiologyThe Muddle of Myoclonus: Many Guises, 2 Disciplines, Consensus Needed.
Neurology. Clinical practiceFamilial Adult Myoclonic Epilepsy: Clinical and Genetic Approach to an Under-recognized Disease.
Noro psikiyatri arsiviCortico-muscular coherence and brain networks in familial adult myoclonic epilepsy and progressive myoclonic epilepsy.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyCurrent treatment options for familial adult myoclonus epilepsy.
EpilepsiaStructures and conformational dynamics of DNA minidumbbells in pyrimidine-rich repeats associated with neurodegenerative diseases.
Computational and structural biotechnology journalDifferential diagnosis of familial adult myoclonic epilepsy.
EpilepsiaA novel FAME1 repeat configuration in a European family identified using a combined genomics approach.
Epilepsia openSolution Nuclear Magnetic Resonance Structures of ATTTT and ATTTC Pentanucleotide Repeats Associated with SCA37 and FAMEs.
ACS chemical neuroscienceAbnormal sensorimotor cortex and thalamo-cortical networks in familial adult myoclonic epilepsy type 2: pathophysiology and diagnostic implications.
Brain communicationsMolecular Mechanisms in Pentanucleotide Repeat Diseases.
CellsFamilial adult myoclonic epilepsy (FAME): clinical features, molecular characteristics, pathophysiological aspects and diagnostic work-up.
Medizinische Genetik : Mitteilungsblatt des Berufsverbandes Medizinische Genetik e.VFamilial adult myoclonic epilepsy type 1 SAMD12 TTTCA repeat expansion arose 17,000 years ago and is present in Sri Lankan and Indian families.
European journal of human genetics : EJHGUnstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3.
Nature communicationsIntronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2.
Nature communicationsFamilial adult myoclonic epilepsy: A new expansion repeats disorder.
SeizureAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Epilepsia mioclônica do adulto familiar.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Epilepsia mioclônica do adulto familiar
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Embryonic spinocerebellar ataxia type 37 AUUUC repeat RNA causes neurodevelopmental defects in zebrafish.
- Novel, complex configurations of the MARCHF6 repeat expansion associated with progressive myoclonic epilepsy and familial adult myoclonic epilepsy.
- [Molecular genetics of benign adult familial myoclonus epilepsy].
- Targeted nanopore long-read sequencing panel for the molecular diagnosis of intronic expansion in familial adult myoclonic epilepsy.
- First clinical diagnosis of FAME3 via commercial Long-Read sequencing reveals mosaic repeat expansion in MARCHF6 gene.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:86814(Orphanet)
- MONDO:0019448(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:16758(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56014305(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Epilepsia mioclônica do adulto familiar
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub