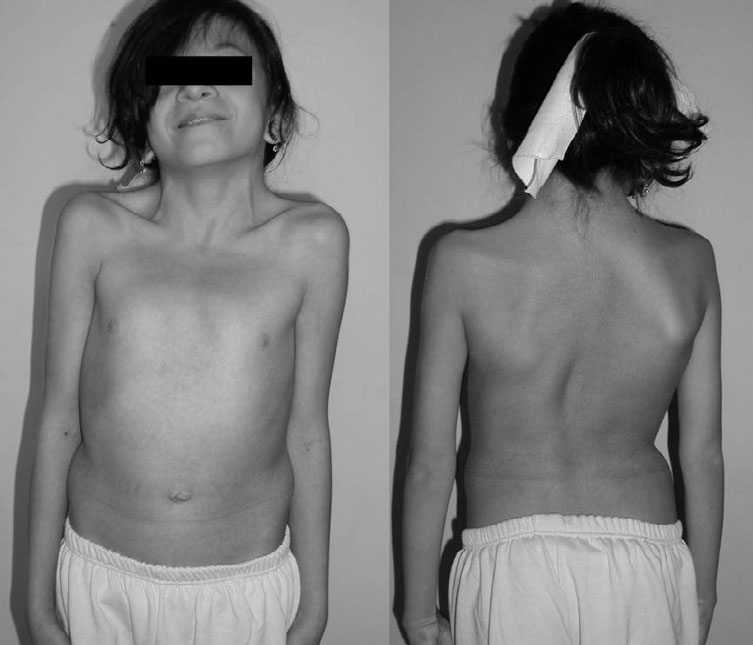
Condição neurológica rara caracterizada principalmente por deficiência intelectual leve a moderada e distonia das mãos. Outros sinais e sintomas podem incluir disartria, anomalias comportamentais, convulsões recorrentes e/ou uma marcha incomum (estilo de andar). A síndrome de Partington geralmente ocorre em homens; quando ocorre em mulheres, os sinais e sintomas costumam ser menos graves. É causada por alterações (mutações) no gene ARX e é herdada de forma recessiva ligada ao X. O tratamento é baseado nos sinais e sintomas presentes em cada pessoa.
Introdução
O que você precisa saber de cara
Condição neurológica rara caracterizada principalmente por deficiência intelectual leve a moderada e distonia das mãos. Outros sinais e sintomas podem incluir disartria, anomalias comportamentais, convulsões recorrentes e/ou uma marcha incomum (estilo de andar). A síndrome de Partington geralmente ocorre em homens; quando ocorre em mulheres, os sinais e sintomas costumam ser menos graves. É causada por alterações (mutações) no gene ARX e é herdada de forma recessiva ligada ao X. O tratamento é baseado nos sinais e sintomas presentes em cada pessoa.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 6 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 19 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: X-linked recessive.
Curadoria gene-doença
fontes oficiaisTranscription factor (PubMed:22194193, PubMed:31691806). Binds to specific sequence motif 5'-TAATTA-3' in regulatory elements of target genes, such as histone demethylase KDM5C (PubMed:22194193, PubMed:31691806). Positively modulates transcription of KDM5C (PubMed:31691806). Activates expression of KDM5C synergistically with histone lysine demethylase PHF8 and perhaps in competition with transcription regulator ZNF711; synergy may be related to enrichment of histone H3K4me3 in regulatory element
Nucleus
Lissencephaly, X-linked 2
A classic type lissencephaly associated with abnormal genitalia. Patients have severe congenital or postnatal microcephaly, lissencephaly, agenesis of the corpus callosum, neonatal-onset intractable epilepsy, poor temperature regulation, chronic diarrhea, and ambiguous or underdeveloped genitalia.
Variantes genéticas (ClinVar)
399 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 18 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Partington
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Managing Dystonia in Partington Syndrome.
Bilateral focal hand dystonia is an almost pathognomonic sign of Partington syndrome, frequently accompanied by intellectual disability and oromotor dyspraxia. However, a few studies have focused on the treatment of this focal dystonia, making patient management uncertain. We present 2 cases of Partington syndrome featuring Aristaless-related homeobox (ARX) gene mutations, hand dystonia, and other clinical signs. Various drug treatments were attempted, including levodopa (l-dopa), trihexyphenidyl, tetrabenazine, and benzodiazepines, as well as botulinum toxin. Additionally, a blinded dystonia protocol was used to assess l-dopa's efficacy in 1 patient, which confirmed only mild benefit. Through a systematic review of the literature, we found that only l-dopa and baclofen might result in mild improvement, whereas propranolol, gabapentin, and haloperidol were reported as ineffective. The descriptions in those studies were, however, imprecise and the improvement rather mild, hindering definitive conclusions about their effectiveness. Treatment options in Partington syndrome-associated dystonia remain elusive. Further research and additional case studies are needed to fully characterize the clinical features of Partington syndrome and to identify effective treatments.
Investigating the "Dark" Genome: First Report of Partington Syndrome in Cyprus.
Background/Objectives: X-linked intellectual disability (XLID) is a highly heterogeneous disorder accounting for ~10% of all males with ID. Next-generation sequencing (NGS) has revolutionized the discovery of causal XLID genes and variants; however, many cases remain unresolved. We present a four-generation syndromic XLID family with multiple males exhibiting variable degree of ID, focal dystonia and epilepsy. Methods: Extensive cytogenetic and targeted genetic testing was initially performed, followed by whole-exome sequencing (WES) and short-read whole-genome sequencing (WGS). Apart from the routine NGS analysis pipelines, sequencing data was revisited by focusing on poorly covered/mapped regions on chromosome X (chrX), to potentially reveal unidentified clinically relevant variants. Candidate variant validation and family segregation analysis were performed with Sanger sequencing. Results: All initial diagnostic testing was negative. Subsequently, 300 previously reported "dark" chrX coding DNA sequences, overlapping 97 genes, were cross-checked against 29 chrX genes highly associated (p < 0.05) with ID and focal dystonia, according to Phenomizer. Manual inspection of the existing NGS data in two low-coverage regions, chrX:25013469-25013696 and chrX:111744737-111744820 (hg38), revealed a recurrent pathogenic ARX variant NM_139058.3:c.441_464dup p.(Ala148_Ala155dup) (ARXdup24) associated with non-syndromic or syndromic XLID, including Partington syndrome. Sanger sequencing confirmed ARXdup24 in all affected males, with carrier status in their unaffected mothers, and absence in other unaffected relatives. Conclusions: After several years of diagnostic odyssey, the pathogenic ARXdup24 variant was unmasked, supporting a genotype-phenotype correlation in the first Partington syndrome family in Cyprus. This study highlights that re-examining underrepresented genomic regions and using phenotype-driven tools can provide critical diagnostic insights in unresolved XLID cases.
Manifestations of Intellectual Disability, Dystonia, and Parkinson's Disease in an Adult Patient with ARX Gene Mutation c.558_560dup p.(Pro187dup).
We describe a 38-year-old male patient with intellectual disability and progressive motor symptoms who lacked an etiological diagnosis for many years. Finally, clinical exome sequencing showed a likely pathogenic variant of the ARX gene suggesting Partington syndrome. His main symptoms were mild intellectual disability, severe kinetic apraxia, resting and action tremor, dysarthria, tonic pupils, constant dystonia of one upper limb, and focal dystonia in different parts of the body, axial rigidity, spasticity, epilepsy, and poor sleep. Another likely pathogenic gene variant was observed in the PKP2 gene and is in accordance with the observed early cardiomyopathy. Single-photon emission computed tomography imaging of dopamine transporters showed a reduced signal in the basal ganglia consistent with Parkinson's disease. Therapies with a variable number of drugs, including antiparkinsonian medications, have yielded poor responses. Our case report extends the picture of the adult phenotype of Partington syndrome.
A comprehensive clinical and genetic study in 127 patients with ID in Kinshasa, DR Congo.
Pathogenic variants account for 4 to 41% of patients with intellectual disability (ID) or developmental delay (DD). In Sub-Saharan Africa, the prevalence of ID is thought to be higher, but data in Central Africa are limited to some case reports. In addition, clinical descriptions of some syndromes are not available for this population. This study aimed at providing an estimate for the fraction of ID/DD for which an underlying etiological genetic cause may be elucidated and provide insights into their clinical presentation in special institutions in a Central African country. A total of 127 patients (33 females and 94 males, mean age 10.03 ± 4.68 years), were recruited from six institutions across Kinshasa. A clinical diagnosis was achieved in 44 but molecular confirmation was achieved in 21 of the 22 patients with expected genetic defect (95% clinical sensitivity). Identified diseases included Down syndrome (15%), submicroscopic copy number variants (9%), aminoacylase deficiency (0.8%), Partington syndrome in one patient (0.8%) and his similarly affected brother, X-linked syndromic Mental Retardation type 33 (0.8%), and two conditions without clear underlying molecular genetic etiologies (Oculo-Auriculo-Vertebral and Amniotic Bands Sequence). We have shown that genetic etiologies, similar to those reported in Caucasian subjects, are a common etiologic cause of ID in African patients from Africa. We have confirmed the diagnostic utility of clinical characterization prior to genetic testing. Finally, our clinical descriptions provide insights into the presentation of these genetic diseases in African patients.
Publicações recentes
Investigating the "Dark" Genome: First Report of Partington Syndrome in Cyprus.
Managing Dystonia in Partington Syndrome.
Manifestations of Intellectual Disability, Dystonia, and Parkinson's Disease in an Adult Patient with ARX Gene Mutation c.558_560dup p.(Pro187dup).
A comprehensive clinical and genetic study in 127 patients with ID in Kinshasa, DR Congo.
Mosaicism for c.431_454dup in ARX causes a mild Partington syndrome phenotype.
📚 EuropePMC5 artigos no totalmostrando 4
Investigating the "Dark" Genome: First Report of Partington Syndrome in Cyprus.
GenesManaging Dystonia in Partington Syndrome.
Movement disorders clinical practiceManifestations of Intellectual Disability, Dystonia, and Parkinson's Disease in an Adult Patient with ARX Gene Mutation c.558_560dup p.(Pro187dup).
Case reports in geneticsA comprehensive clinical and genetic study in 127 patients with ID in Kinshasa, DR Congo.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Partington.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Partington
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Managing Dystonia in Partington Syndrome.
- Investigating the "Dark" Genome: First Report of Partington Syndrome in Cyprus.
- Manifestations of Intellectual Disability, Dystonia, and Parkinson's Disease in an Adult Patient with ARX Gene Mutation c.558_560dup p.(Pro187dup).
- A comprehensive clinical and genetic study in 127 patients with ID in Kinshasa, DR Congo.
- Mosaicism for c.431_454dup in ARX causes a mild Partington syndrome phenotype.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:94083(Orphanet)
- OMIM OMIM:309510(OMIM)
- MONDO:0010654(MONDO)
- GARD:4235(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18554811(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Partington
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata